Acta Chimica Sinica ›› 2022, Vol. 80 ›› Issue (7): 888-895.DOI: 10.6023/A22030109 Previous Articles Next Articles
Article
李海茹a,*( ), 张层b, 李思殿c
), 张层b, 李思殿c
投稿日期:2022-03-15
发布日期:2022-05-05
通讯作者:
李海茹
基金资助:
Hairu Lia(), Ceng Zhangb, Sidian Lic
Received:2022-03-15
Published:2022-05-05
Contact:
Hairu Li
Supported by:Share
Hairu Li, Ceng Zhang, Sidian Li. Study on the Regulation of Alkali-earth Metal Ben (n=1~3) on the Structure of B12 Clusters[J]. Acta Chimica Sinica, 2022, 80(7): 888-895.
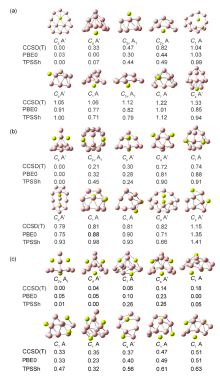


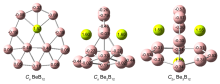
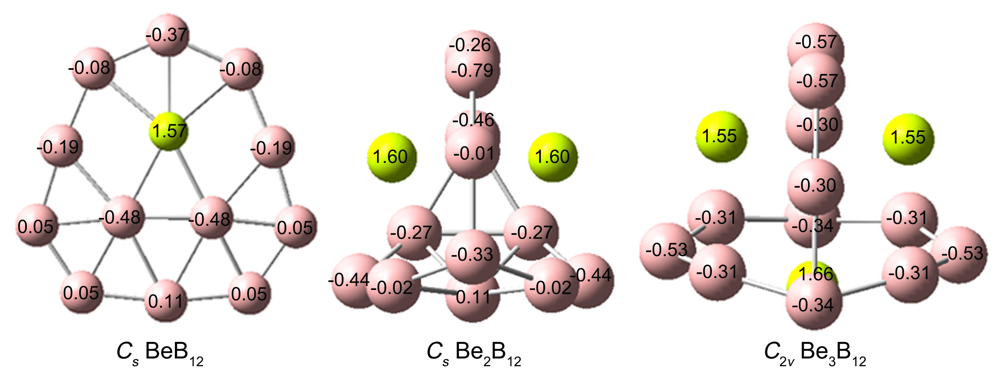
| BeB12 | Be2B12 | Be3B12 | |
|---|---|---|---|
| B—B键长范围 | 0.155~0.183 | 0.153~0.187 | 0.151~0.196 |
| B—Be键长范围 | 0.185~0.206 | 0.191~0.225 | 0.188~0.216 |
| Be—Be键长范围 | — | 0.240 | 0.261~0.269 |
| Be—B WBIs | 0.01~0.17 | 0.01~0.15 | 0.02~0.17 |
| Be—Be WBIs | 0.00 | 0.00~0.01 | 0.00~0.01 |
| B价电子构型 | 2s0.65–0.932p2.05–2.81 | 2s0.60–0.892p2.09–2.86 | 2s0.80–0.872p2.44–2.67 |
| Be价电子构型 | 2s0.212p0.19 | 2s0.222p0.15 | 2s0.16–0.282p0.15–0.16 |
| BeB12 | Be2B12 | Be3B12 | |
|---|---|---|---|
| B—B键长范围 | 0.155~0.183 | 0.153~0.187 | 0.151~0.196 |
| B—Be键长范围 | 0.185~0.206 | 0.191~0.225 | 0.188~0.216 |
| Be—Be键长范围 | — | 0.240 | 0.261~0.269 |
| Be—B WBIs | 0.01~0.17 | 0.01~0.15 | 0.02~0.17 |
| Be—Be WBIs | 0.00 | 0.00~0.01 | 0.00~0.01 |
| B价电子构型 | 2s0.65–0.932p2.05–2.81 | 2s0.60–0.892p2.09–2.86 | 2s0.80–0.872p2.44–2.67 |
| Be价电子构型 | 2s0.212p0.19 | 2s0.222p0.15 | 2s0.16–0.282p0.15–0.16 |
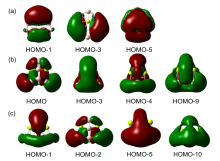




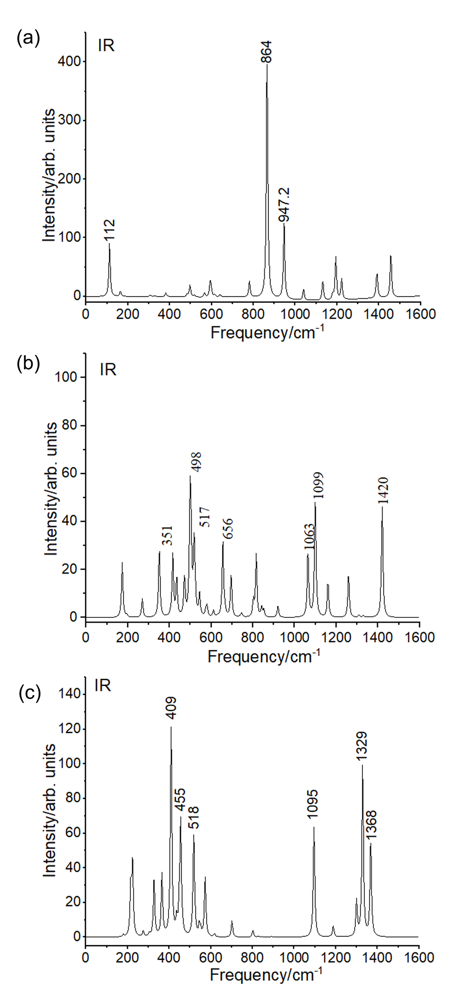
| [1] |
Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162.
doi: 10.1038/318162a0 |
| [2] |
Krätschmer, W.; Lamb, L. D.; Fostiropoulos, K.; Huffman, D. R. Nature 1990, 347, 354.
doi: 10.1038/347354a0 |
| [3] |
Iijima, S. Nature 1991, 354, 56.
doi: 10.1038/354056a0 |
| [4] |
Iijima, S.; Ichihashi, T. Nature 1993, 363, 603.
doi: 10.1038/363603a0 |
| [5] |
Alexandrova, A. N.; Boldyrev, A. I.; Zhai, H. J.; Wang, L. S. Coordin. Chem. Rev. 2006, 250, 2811.
doi: 10.1016/j.ccr.2006.03.032 |
| [6] |
Sergeeva, A. P.; Popov, I. A.; Piazza, Z. A.; Li, W. L.; Romanescu, C.; Wang, L. S.; Boldyrev, Alexander I. Acc. Chem. Res. 2014, 47, 1349.
doi: 10.1021/ar400310g |
| [7] |
Wang, L. S. Int. Rev. Phys. Chem. 2016, 35, 69.
doi: 10.1080/0144235X.2016.1147816 |
| [8] |
Zhai, H. J.; Zhao, Y. F.; Li, W. L.; Chen, Q.; Bai, H.; Hu, H. S.; Piazza, Z. A.; Tian, W. J.; Lu, H. G.; Wu, Y. B.; Mu, Y. W.; Wei, G. F.; Liu, Z. P.; Li, J.; Li, S. D.; Wang, L. S. Nat. Chem. 2014, 6, 727.
doi: 10.1038/nchem.1999 |
| [9] |
Chen, Q.; Li, H. R.; Tian, W. J.; Lu, H. G.; Zhai, H. J.; Li, S. D. Phys. Chem. Chem. Phys. 2016, 18, 14186.
doi: 10.1039/C6CP02369J |
| [10] |
Romanescu, C.; Galeev, T. R.; Li, W. L.; Boldyrev, A. I.; Wang, L. S. Angew. Chem., Int. Ed. 2011, 50, 9334.
doi: 10.1002/anie.201104166 |
| [11] |
Li, W. L.; Romanescu, C.; Galeev, T. R.; Piazza, Z. A.; Boldyrev, A. I.; Wang, L. S. J. Am. Chem. Soc. 2012, 134, 165-168.
doi: 10.1021/ja209808k |
| [12] |
Galeev, T. R.; Romanescu, C.; Li, W. L.; Wang, L. S.; Boldyrev, A. I. Angew. Chem., Int. Ed. 2012, 51, 2101.
doi: 10.1002/anie.201107880 pmid: 22298320 |
| [13] |
Romanescu, C.; Galeev, T. R.; Li, W. L.; Boldyrev, A. I.; Wang, L. S. J. Chem. Phys. 2013, 138, 134315.
doi: 10.1063/1.4798935 |
| [14] |
Li, W. L.; Ivanov, A. S.; Federi, J.; Romanescu, C.; ernušák, I.; Boldyrev, A. I.; Wang, L. S. J. Chem. Phys. 2013, 139, 104312.
doi: 10.1063/1.4820401 |
| [15] |
Romanescu, C.; Galeev, T. R.; Li, W. L.; Boldyrev, A. I.; Wang, L. S. Acc. Chem. Res. 2013, 46, 350.
doi: 10.1021/ar300149a |
| [16] |
Popov, I. A.; Li, W. L.; Piazza, Z. A.; Boldyrev, A. I.; Wang, L. S. J. Phys. Chem. A 2014, 118, 8098.
doi: 10.1021/jp411867q pmid: 24428747 |
| [17] |
Popov, I. A.; Jian, T.; Lopez, G. V.; Boldyrev, A. I.; Wang, L. S. Nat. Commun. 2015, 6, 8654.
doi: 10.1038/ncomms9654 pmid: 26456760 |
| [18] |
Jian, T.; Li, W. L.; Popov, I. A.; Lopez, G. V.; Chen, X.; Boldyrev, A. I.; Li, J.; Wang, L. S. J. Chem. Phys. 2016, 144, 154310.
doi: 10.1063/1.4946796 |
| [19] |
Jian, T.; Li, W. L.; Chen, X.; Chen, T. T.; Lopez, G. V.; Li, J.; Wang, L. S. Chem. Sci. 2016, 7, 7020.
doi: 10.1039/C6SC02623K |
| [20] |
Li, W. L.; Jian, T.; Chen, X.; Chen, T. T.; Lopez, G. V.; Li, J.; Wang, L. S. Angew. Chem., Int. Ed. 2016, 55, 7358.
doi: 10.1002/anie.201601548 |
| [21] |
Li, H. R.; Liu, H.; Tian, X. X.; Zan, W. Y.; Mu, Y. W.; Lu, H. G.; Li, J.; Wang, Y. K.; Li, S. D. Phys. Chem. Chem. Phys. 2017, 19, 27025.
doi: 10.1039/C7CP05179D |
| [22] |
Li, H. R.; Liu, H.; Lu, X. Q.; Zan, W. Y.; Tian, X. X.; Lu, H. G.; Wu, Y. B.; Mu, Y. W.; Li, S. D. Nanoscale 2018, 10, 7451.
doi: 10.1039/C8NR01087K |
| [23] |
Li, H. R. Ph.D. Dissertation, Shanxi University, Taiyuan, 2019. (in Chinese)
|
|
(李海茹, 博士论文, 山西大学, 太原, 2019.)
|
|
| [24] |
Wang, Y. J.; Feng, L. Y.; Guo, J. C.; Zhai, H. J. Chem-Asian J. 2017, 12, 2899.
doi: 10.1002/asia.201701310 |
| [25] |
Li, S. X.; Chen, D. L.; Zhang, Z. L.; Long, Z. W. Acta Phys. Sin. 2020, 69, 193101. (in Chinese)
doi: 10.7498/aps.69.20200756 |
|
(李世雄, 陈德良, 张正良, 隆正文, 物理学报, 2020, 69, 193101.)
|
|
| [26] |
Dong, X.; Jalife, S.; Vásquez-Espinal, A.; Ravell, E.; Pan, S.; Cabellos, J. L.; Liang, W. Y.; Cui, Z. H.; Merino, G. Angew. Chem. 2018, 130, 1.
doi: 10.1002/ange.201712460 |
| [27] |
Feng, L. Y. Ph.D. Dissertation, Shanxi University, Taiyuan, 2021. (in Chinese)
|
|
(冯林雁, 博士论文, 山西大学, 太原, 2021.)
|
|
| [28] |
Cui, Z. H.; Yang, W. S.; Zhao, L. L.; Ding, Y. H.; Frenking, G. Angew. Chem., Int. Ed., 2016, 55, 7841.
doi: 10.1002/anie.201601890 |
| [29] |
Feng, L. Y.; Guo, J. C.; Li, P. F.; Zhai, H. J. Phys. Chem. Chem Phys. 2018, 20, 22719.
doi: 10.1039/C8CP04332A |
| [30] |
Guo, J. C.; Feng, L. Y.; Wang, Y. J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J. L.; Pan, S.; Merino, G.; Zhai, H. J. Angew. Chem., Int. Ed. 2017, 56, 10174.
doi: 10.1002/anie.201703979 |
| [31] |
Feng, L. Y.; Wang, K.; Zhai, H. J. Phys. Chem. Chem. Phys. 2020, 22, 25574.
doi: 10.1039/D0CP05012A |
| [32] |
Feng, L. Y.; Guo, J. C.; Li, P. F.; Zhai, H. J. Chem. Asian J. 2020, 15, 1094.
doi: 10.1002/asia.201901640 |
| [33] |
Zhai, H. J.; Kiran, B.; Li, J.; Wang, L. S. Nat. Mater. 2003, 2, 827.
doi: 10.1038/nmat1012 |
| [34] |
Zhao, Y. F.; Chen, X.; Li, J. Nano Research. 2017, 10, 3407.
doi: 10.1007/s12274-017-1553-z |
| [35] |
Chen, X.; Zhao, Y. F.; Wang, L. S.; Li, J. Comput. Theor. Chem. 2017, 1107, 57.
doi: 10.1016/j.comptc.2016.12.028 |
| [36] |
Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158.
doi: 10.1063/1.478522 |
| [37] |
Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Phys. Rev. Lett. 2003, 91, 146401.
doi: 10.1103/PhysRevLett.91.146401 |
| [38] |
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980. 72, 650.
|
| [39] |
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16 Revision, A. 03, Gaussian Inc., Wallingford, CT, 2016.
|
| [40] |
Zubarev, D. Y.; Boldyrev, A. I. Phys. Chem. Chem. Phys. 2008, 10, 5207.
doi: 10.1039/b804083d pmid: 18728862 |
| [41] |
Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Bohmann, J. A.; Morales, C. M.; Weinhold, F.NBO 5.0/6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001.
|
| [42] |
Vondele, J. V.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Comput. Phys. Commun. 2005, 167, 103.
doi: 10.1016/j.cpc.2004.12.014 |
| [43] |
Pyykkö, P. J. Phys. Chem. A. 2015, 119, 2326.
doi: 10.1021/jp5065819 pmid: 25162610 |
| [44] |
Li, S. D.; Zhai, H. J.; Wang, L. S. J. Am. Chem. Soc. 2008, 130, 2573.
doi: 10.1021/ja0771080 |
| [45] |
Kalemos, A. J. Chem. Phys. 2016, 145, 214302.
doi: 10.1063/1.4967819 |
| [46] |
Wang, G.; Zhou, M.; Goettel, J. T.; Schrobilgen, G. J.; Su, J.; Li, J.; Schlöder, T.; Riedel, S. Nature 2014, 514, 475.
doi: 10.1038/nature13795 |
| [1] | Zhao Zigang, Niu Yongqiang, Zhao Yang, Song Qinghua, Xin Ling, Lu Xiaoqing. First-Principles Theory Investigation on Structural and Photoelectronic Properties of Formamidinium Lead Halide Perovskites [J]. Acta Chim. Sinica, 2016, 74(8): 689-693. |
| [2] | Zhao Zigang, Lu Xiaoqing, Li Ke, Wei Shuxian, Liu Xuefeng, Niu Kai, Guo Wenyue. First-Principles Theory Investigation on Structural and Photoelectronic Properties of Perovskites:Trigonal versus Hexagonal HC(NH2)2PbI3 [J]. Acta Chim. Sinica, 2016, 74(12): 1003-1008. |
| [3] | Yu Hui, Guo Juan, Wang Jing, Long Yingcai. Growth Mechanism Study on b-axis Oriented MFI Zeolite with MD Simulation [J]. Acta Chimica Sinica, 2012, 70(14): 1543-1550. |
| [4] | JIA Xue-Bing, ZHANG Jun, SUN Hong-Wei, CHEN Lan, SHEN Rong-Xin, LAI Cheng-Ming. Molecular Dynamics Simulation Study on the Interaction Mode of Auxin Perception [J]. Acta Chimica Sinica, 2010, 68(24): 2500-2508. |
| [5] | SHANG Wei, WANG Wen-Ju, WANG Song. DFT Calculations on the Adsorption of Small Gas Molecules onto Pt-doped Carbon Nanotubes [J]. Acta Chimica Sinica, 2010, 68(23): 2389-2394. |
| [6] | PAN Yong-Mei, HOU Ting-Jun, JI Ming-Juan, XU Xiao-Jie, YE Xue-Qi. MD Simulations of PTP1B-Inhibitor Complex [J]. Acta Chimica Sinica, 2004, 62(2): 148-152. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
