Acta Chimica Sinica ›› 2022, Vol. 80 ›› Issue (10): 1401-1409.DOI: 10.6023/A22070313 Previous Articles Next Articles
Article
王治钒a,*( ), 何冰a, 路艳朝b, 王繁b
), 何冰a, 路艳朝b, 王繁b
投稿日期:2022-07-17
发布日期:2022-08-17
通讯作者:
王治钒
基金资助:
Zhifan Wanga(), Bing Hea, Yanzhao Lub, Fan Wangb
Received:2022-07-17
Published:2022-08-17
Contact:
Zhifan Wang
Supported by:Share
Zhifan Wang, Bing He, Yanzhao Lu, Fan Wang. Single-precision CCSD and CCSD(T) Calculations with Density Fitting Approximations on Graphics Processing Units[J]. Acta Chimica Sinica, 2022, 80(10): 1401-1409.



| CCSD | (T) | ||||||
|---|---|---|---|---|---|---|---|
| Molecular | SP | SP | DP | SP | SP | DP | |
| GPU | CPU | CPU | GPU | CPU | CPU | ||
| (H2O)7 | –1.9878328 | –1.9878329 | –1.9878329 | –0.0585549 | –0.0585566 | –0.0585566 | |
| C6H14 | –1.1398594 | –1.1398593 | –1.1398594 | –0.0440448 | –0.0440449 | –0.0440449 | |
| C10H16 | –1.7801039 | –1.7801038 | –1.7801038 | –0.0798821 | –0.0798823 | –0.0798823 | |
| AtF5 | –2.2048644 | –2.2048637 | –2.2048637 | –0.0941050 | –0.0940953 | –0.0941050 | |
| I3- | –1.3032696 | –1.3032697 | –1.3032697 | –0.0498108 | –0.0498108 | –0.0498108 | |
| CCSD | (T) | ||||||
|---|---|---|---|---|---|---|---|
| Molecular | SP | SP | DP | SP | SP | DP | |
| GPU | CPU | CPU | GPU | CPU | CPU | ||
| (H2O)7 | –1.9878328 | –1.9878329 | –1.9878329 | –0.0585549 | –0.0585566 | –0.0585566 | |
| C6H14 | –1.1398594 | –1.1398593 | –1.1398594 | –0.0440448 | –0.0440449 | –0.0440449 | |
| C10H16 | –1.7801039 | –1.7801038 | –1.7801038 | –0.0798821 | –0.0798823 | –0.0798823 | |
| AtF5 | –2.2048644 | –2.2048637 | –2.2048637 | –0.0941050 | –0.0940953 | –0.0941050 | |
| I3- | –1.3032696 | –1.3032697 | –1.3032697 | –0.0498108 | –0.0498108 | –0.0498108 | |
| Wall-time of a single RI-CCSD iteration | Wall-time of (T) correction calculation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SP | SP | SP | DP | SP | SP | SP | DP | |||
| n | Number of basis | GPU Time | CPU Time | $\frac{\text{CPUTime}}{\text{GPUTime}}$ | CPU Time | GPU Time | CPU Time | $\frac{\text{CPUTime}}{\text{GPUTime}}$ | CPU Time | |
| 3 | 174 | 0.9 (0.4) | 5.46 (2.24) | 6.07 (5.6) | 12.4 (5.2) | 2.8 (1.8) | 22.4 (13.9) | 8 (7.7) | 40 (33) | |
| 4 | 232 | 3.1 (1.4) | 17.7 (9.3) | 5.71 (6.64) | 37.2 (19.0) | 14 (6.7) | 336 (105) | 24 (15.7) | 672 (244) | |
| 5 | 290 | 7.8 (3.7) | 50.9 (24.6) | 6.53 (6.65) | 106 (52.1) | 62 (23) | 1514 (492) | 24.4 (21.4) | 2698 (1035) | |
| 6 | 348 | 18 (8) | 129 (58) | 7.17 (7.25) | 243 (122) | 189 (69) | 4866 (1555) | 25.7 (22.5) | 8476 (3174) | |
| 7 | 406 | 35 (14) | 283 (111) | 8.09 (7.93) | 523 (226) | 542 (179) | 12588 (4359) | 23.2 (24.4) | 22312 (8399) | |
| 8 | 464 | 66 (28) | 508 (220) | 7.7 (7.86) | 1012 (436) | 1514 (445) | 31404 (11527) | 20.7 (25.9) | 53604 (19188) | |
| 9 | 522 | 131 (44) | 913 (389) | 6.97 (8.84) | 1815 (774) | 3601 (898) | 65888 (22203) | 18.3 (24.7) | 113226 (40485) | |
| 10 | 580 | 529 (81) | 1768 (656) | 3.34 (8.1) | (1626) | 7608 (2313) | 119307 (46606) | 15.7 (20.2) | (84115) | |
| Wall-time of a single RI-CCSD iteration | Wall-time of (T) correction calculation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SP | SP | SP | DP | SP | SP | SP | DP | |||
| n | Number of basis | GPU Time | CPU Time | $\frac{\text{CPUTime}}{\text{GPUTime}}$ | CPU Time | GPU Time | CPU Time | $\frac{\text{CPUTime}}{\text{GPUTime}}$ | CPU Time | |
| 3 | 174 | 0.9 (0.4) | 5.46 (2.24) | 6.07 (5.6) | 12.4 (5.2) | 2.8 (1.8) | 22.4 (13.9) | 8 (7.7) | 40 (33) | |
| 4 | 232 | 3.1 (1.4) | 17.7 (9.3) | 5.71 (6.64) | 37.2 (19.0) | 14 (6.7) | 336 (105) | 24 (15.7) | 672 (244) | |
| 5 | 290 | 7.8 (3.7) | 50.9 (24.6) | 6.53 (6.65) | 106 (52.1) | 62 (23) | 1514 (492) | 24.4 (21.4) | 2698 (1035) | |
| 6 | 348 | 18 (8) | 129 (58) | 7.17 (7.25) | 243 (122) | 189 (69) | 4866 (1555) | 25.7 (22.5) | 8476 (3174) | |
| 7 | 406 | 35 (14) | 283 (111) | 8.09 (7.93) | 523 (226) | 542 (179) | 12588 (4359) | 23.2 (24.4) | 22312 (8399) | |
| 8 | 464 | 66 (28) | 508 (220) | 7.7 (7.86) | 1012 (436) | 1514 (445) | 31404 (11527) | 20.7 (25.9) | 53604 (19188) | |
| 9 | 522 | 131 (44) | 913 (389) | 6.97 (8.84) | 1815 (774) | 3601 (898) | 65888 (22203) | 18.3 (24.7) | 113226 (40485) | |
| 10 | 580 | 529 (81) | 1768 (656) | 3.34 (8.1) | (1626) | 7608 (2313) | 119307 (46606) | 15.7 (20.2) | (84115) | |
| (H2O)9 | (H2O)10 | ||||
|---|---|---|---|---|---|
| GPU | CPU | GPU | CPU | ||
| 1) Initio | 15.6 | 15.3 | 233.7 | 235 | |
| 2) N5 | 60 | 53.6 | 84.5 | 87.1 | |
| 3) Iabci | 3.5 | 22 | 7 | 37.6 | |
| 4) Iabcd | 37.7 | 623 | 66 | 1050 | |
| 5) Wmbej | 10.79 | 169.9 | 320.2 | 319.2 | |
| 6) left | 2.5 | 18.9 | 3.5 | 30 | |
| (H2O)9 | (H2O)10 | ||||
|---|---|---|---|---|---|
| GPU | CPU | GPU | CPU | ||
| 1) Initio | 15.6 | 15.3 | 233.7 | 235 | |
| 2) N5 | 60 | 53.6 | 84.5 | 87.1 | |
| 3) Iabci | 3.5 | 22 | 7 | 37.6 | |
| 4) Iabcd | 37.7 | 623 | 66 | 1050 | |
| 5) Wmbej | 10.79 | 169.9 | 320.2 | 319.2 | |
| 6) left | 2.5 | 18.9 | 3.5 | 30 | |
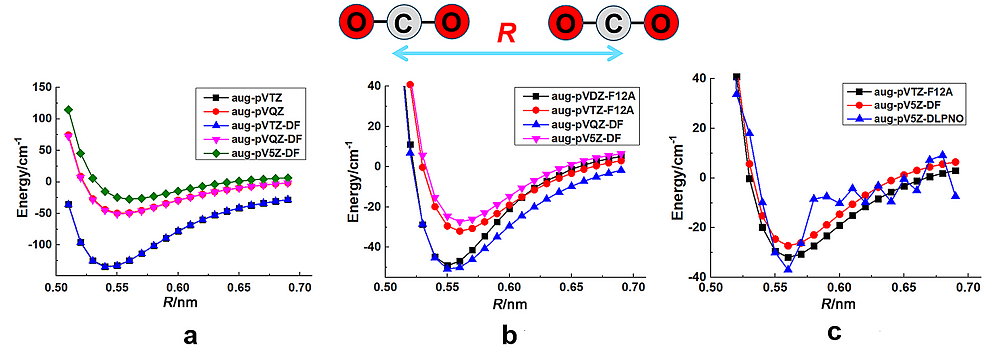
| Reaction | cc-pVTZ CCSD(T) | cc-pVTZ DF- CCSD(T) | cc-pVTZ DLPNO- CCSD(T) | cc-pVQZ DF- CCSD(T) | cc-pVQZ DLPNO- CCSD(T) | cc-pVDZ CCSD(T)- F12a | cc-pV5Z DF- CCSD(T) | cc-pV5Z DLPNO- CCSD(T) | cc-pVTZ CCSD(T)- F12a | Ref. a |
|---|---|---|---|---|---|---|---|---|---|---|
| (1) E22→E1 | 56.028 | 55.823 | 56.551 | 53.145 | 57.618 | 85.713 | 59.865 | 66.35 | 59.999 | |
| (2) E31→E1 | 94.847 | 94.508 | 94.583 | 86.768 | 95.42 | 133.955 | 97.345 | 107.106 | 104.684 | |
| (3) (CH3)3CC(CH3)3→C8H18 | 8.694 | 8.715 | 6.904 | 7.736 | 6.464 | 9.301 | 6.209 | 8.556 | 7.95 | |
| (4) C6H14+4CH4→5C2H6 | 35.204 | 35.179 | 33.359 | 36.53 | 34.623 | 35.539 | 38.777 | 35.585 | 35.514 | 41.045 |
| (5) C8H18+6CH4→7C2H6 | 53.099 | 53.057 | 49.773 | 55.078 | 51.953 | 53.321 | 58.501 | 53.597 | 53.48 | 62.091 |
| (6) adamantane→3C2H4+2C2H2 | 819.855 | 818.754 | 817.298 | 821.675 | 820.608 | 890.255 | 822.089 | 825.968 | 840.009 | 811.654 |
| (7) bicyclo_octane→3C2H4+C2H2 | 530.996 | 530.251 | 527.661 | 532.824 | 530.464 | 581.706 | 531.841 | 535.088 | 547.024 | 532.288 |
| Reaction | cc-pVTZ CCSD(T) | cc-pVTZ DF- CCSD(T) | cc-pVTZ DLPNO- CCSD(T) | cc-pVQZ DF- CCSD(T) | cc-pVQZ DLPNO- CCSD(T) | cc-pVDZ CCSD(T)- F12a | cc-pV5Z DF- CCSD(T) | cc-pV5Z DLPNO- CCSD(T) | cc-pVTZ CCSD(T)- F12a | Ref. a |
|---|---|---|---|---|---|---|---|---|---|---|
| (1) E22→E1 | 56.028 | 55.823 | 56.551 | 53.145 | 57.618 | 85.713 | 59.865 | 66.35 | 59.999 | |
| (2) E31→E1 | 94.847 | 94.508 | 94.583 | 86.768 | 95.42 | 133.955 | 97.345 | 107.106 | 104.684 | |
| (3) (CH3)3CC(CH3)3→C8H18 | 8.694 | 8.715 | 6.904 | 7.736 | 6.464 | 9.301 | 6.209 | 8.556 | 7.95 | |
| (4) C6H14+4CH4→5C2H6 | 35.204 | 35.179 | 33.359 | 36.53 | 34.623 | 35.539 | 38.777 | 35.585 | 35.514 | 41.045 |
| (5) C8H18+6CH4→7C2H6 | 53.099 | 53.057 | 49.773 | 55.078 | 51.953 | 53.321 | 58.501 | 53.597 | 53.48 | 62.091 |
| (6) adamantane→3C2H4+2C2H2 | 819.855 | 818.754 | 817.298 | 821.675 | 820.608 | 890.255 | 822.089 | 825.968 | 840.009 | 811.654 |
| (7) bicyclo_octane→3C2H4+C2H2 | 530.996 | 530.251 | 527.661 | 532.824 | 530.464 | 581.706 | 531.841 | 535.088 | 547.024 | 532.288 |

| [1] |
Bartlett, R. J.; Musiał, M. Rev. Mod. Phys. 2007, 79, 291.
doi: 10.1103/RevModPhys.79.291 |
| [2] |
Raghavachari, K.; Trucks, G. W.; Pople, J. A.; Head-Gordon, M. Chem. Phys. Lett. 1989, 157, 479.
doi: 10.1016/S0009-2614(89)87395-6 |
| [3] |
Řezáč, J.; Hobza, P. J. Chem. Theor. Comput. 2013, 9, 2151.
doi: 10.1021/ct400057w |
| [4] |
(a) Vahtras, O.; Almlöf, J.; Feyereisen, M. W. Chem. Phys. Lett. 1993, 213, 514.
doi: 10.1016/0009-2614(93)89151-7 |
|
(b) Van Alsenoy, C. J. Comput. Chem. 1988, 9, 620.
doi: 10.1002/jcc.540090607 |
|
| [5] |
(a) Kats, D.; Korona, T.; Schütz, M. J. Chem. Phys. 2007, 127, 064107.
doi: 10.1063/1.2755778 |
|
(b) Korona, T.; Jeziorski, B. J. Chem. Phys. 2008, 128, 144107.
doi: 10.1063/1.2889006 |
|
|
(c) Boström, J.; Pitoňák, M.; Aquilante, F.; Neogrády, P.; Pedersen, T. B.; Lindh, R. J. Chem. Theor. Comput. 2012, 8, 1921.
doi: 10.1021/ct3003018 |
|
|
(d) DePrince, A. E.; Sherrill, C. D. J. Chem. Theor. Comput. 2013, 9, 2687.
doi: 10.1021/ct400250u |
|
|
(e) Epifanovsky, E.; Zuev, D.; Feng, X.; Khistyaev, K.; Shao, Y.; Krylov, A. I. J. Chem. Phys. 2013, 139, 134105.
doi: 10.1063/1.4820484 |
|
|
(f) DePrince, A. E.; Kennedy, M. R.; Sumpter, B. G.; Sherrill, C. D. Mol. Phys. 2014, 112, 844.
doi: 10.1080/00268976.2013.874599 |
|
|
(g) Lesiuk, M. J. Chem. Phys. 2020, 152, 044104.
doi: 10.1063/1.5129883 |
|
|
(h) Bozkaya, U.; Sherrill, C. D. J. Chem. Phys. 2017, 147, 044104.
doi: 10.1063/1.4994918 |
|
|
(i) Shen, T.; Zhu, Z.; Zhang, I. Y.; Scheffler, M. J. Chem. Theor. Comput. 2019, 15, 4721.
doi: 10.1021/acs.jctc.8b01294 |
|
| [6] |
(a) Harvey, M. J.; De Fabritiis, G. WIREs: Comput. Mol. Sci. 2012, 2, 734.
doi: 10.1002/wcms.1101 |
|
(b) Walker, R. C.; Götz, A. W. Electronic Structure Calculations on Graphics Processing Units. (Wiley Online Library, 2016).
|
|
|
(c) Bao, J. Z.; Feng, X. T.; Yu, J. G. Acta Phys. Chim. Sin. 2011, 27, 2019. (in Chinese)
doi: 10.3866/PKU.WHXB20110830 |
|
|
(鲍建樟, 丰鑫田, 于建国, 物理化学学报, 2011, 27, 2019.)
|
|
| [7] |
(a) Stopper, D.; Roth, R. J. Chem. Phys. 2017, 147, 064508.
doi: 10.1063/1.4997636 |
|
(b) Nitsche, M. A.; Ferreria, M.; Mocskos, E. E.; Gonzalez Lebrero, M. C. J. Chem. Theor. Comput. 2014, 10, 959.
doi: 10.1021/ct400308n |
|
|
(c) Jia, W.; Fu, J.; Cao, Z.; Wang, L.; Chi, X.; Gao, W.; Wange, L. W. J. Comput. Phys. 2013, 251, 102.
|
|
|
(d) Genovese, L.; Ospici, M.; Deutsch, T.; Mehaut, J. F.; Neelov, A.; Goedecker, S. J. Chem. Phys. 2009, 131, 034103.
doi: 10.1063/1.3166140 |
|
| [8] |
(a) Tornai, G. J.; Ladjanszki, I.; Rak, A.; Kis, G.; Cserey, G. J. Chem. Theor. Comput. 2019, 15, 5319.
doi: 10.1021/acs.jctc.9b00560 |
|
(b) Rak, A.; Cserey, G. Chem. Phys. Lett. 2015, 622, 92.
doi: 10.1016/j.cplett.2015.01.023 |
|
|
(c) Asadchev, A.; Gordon, M. S. J. Chem. Theor. Comput. 2012, 8, 4166.
doi: 10.1021/ct300526w |
|
|
(d) Wang, Y.; Tian, Y. Q.; Jin, Z.; Suo, B. B. Acta Chim. Sinica 2021, 79, 653. (in Chinese)
doi: 10.6023/A21020044 |
|
|
(王岩, 田英齐, 金钟, 索兵兵, 化学学报, 2021, 79, 653.)
doi: 10.6023/A21020044 |
|
| [9] |
(a) Bykov, D.; Kjaergaard, T. J. Comput. Chem. 2017, 38, 228.
doi: 10.1002/jcc.24678 |
|
(b) Angel Martinez-Martinez, L.; Amador-Bedolla, C. J. Mex. Chem. Soc. 2017, 61, 60.
|
|
|
(c) Song, C.; Martinez, T. J. J. Chem. Phys. 2016, 144, 174111.
doi: 10.1063/1.4948438 |
|
|
(d) Maurer, S. A.; Kussmann, J.; Ochsenfeld, C. J. Chem. Phys. 2014, 141, 051106.
doi: 10.1063/1.4891797 |
|
| [10] |
Fales, B. S.; Levine, B. G. J. Chem. Theor. Comput. 2015, 11, 4708.
doi: 10.1021/acs.jctc.5b00634 |
| [11] |
Fales, B. S.; Curtis, E. R.; Johnson, K. G.; Lahana, D.; Seritan, S.; Wang, Y.; Weir, H.; Martínez, T. J.; Hohenstein, E. G. J. Chem. Theor. Comput. 2020, 16, 2021.
doi: 10.1021/acs.jctc.9b01257 |
| [12] |
Knizia, G.; Li, W.; Simon, S.; Werner, H. J. J. Chem. Theor. Comput. 2011, 7, 2387.
doi: 10.1021/ct200239p |
| [13] |
Pokhilko, P.; Epifanovsky, E.; Krylov, A. I. J. Chem. Theor. Comput. 2018, 14, 4088.
doi: 10.1021/acs.jctc.8b00321 |
| [14] |
Wang, Z.; Guo, M.; Wang, F. Phys. Chem. Chem. Phys. 2020, 22, 25103.
doi: 10.1039/D0CP03800H |
| [15] |
Knizia, G.; Adler, T. B.; Werner, H.-J. J. Chem. Phys. 2009, 130, 054104.
doi: 10.1063/1.3054300 |
| [16] |
(a) Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D. G.; Minenkov, Y.; Cavallo, L.; Neese, F. J. Chem. Phys. 2018, 148, 011101.
doi: 10.1063/1.5011798 |
|
(b) Liakos, D. G.; Guo, Y.; Neese, F. J. Phys. Chem. A 2020, 124, 90.
doi: 10.1021/acs.jpca.9b05734 |
|
| [17] |
Gauss, J.; Stanton, J. F.; Bartlett, R. J. J. Chem. Phys. 1991, 95, 2623.
|
| [18] |
(a) Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S.; Wouters, S.; Chan, G. K.-L. WIREs Comput. Mol. Sci. 2018, 8, 1340.
|
|
(b) Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N. S.; Bogdanov, N. A.; Booth, G. H.; Chen, J.; Cui, Z.-H.; Eriksen, J. J.; Gao, Y.; Guo, S.; Hermann, J.; Hermes, M. R.; Koh, K.; Koval, P.; Lehtola, S.; Li, Z.; Liu, J.; Mardirossian, N.; McClain, J. D.; Motta, M.; Mussard, B.; Pham, H. Q.; Pulkin, A.; Purwanto, W.; Robinson, P. J.; Ronca, E.; Sayfutyarova, E. R.; Scheurer, M.; Schurkus, H. F.; Smith, J. E. T.; Sun, C.; Sun, S.-N.; Upadhyay, S.; Wagner, L. K.; Wang, X.; White, A.; Whitfield, J. D.; Williamson, M. J.; Wouters, S.; Yang, J.; Yu, J. M.; Zhu, T.; Berkelbach, T. C.; Sharma, S.; Sokolov, A. Y.; Chan, G. K.-L. J. Chem. Phys. 2020, 153, 024109.
doi: 10.1063/5.0006074 |
|
| [19] |
Dunning, T. H. J. Chem. Phys. 1989, 90, 1007.
doi: 10.1063/1.456153 |
| [20] |
Weigend, F. Phys. Chem. Chem. Phys. 2002, 4, 4285.
doi: 10.1039/b204199p |
| [21] |
(a) Peterson, K. A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. J. Chem. Phys. 2003, 119, 11113.
pmid: 17181347 |
|
(b) Peterson, K. A.; Shepler, B. C.; Figgen, D.; Stoll, H. J. Phys. Chem. A 2006, 110, 13877.
pmid: 17181347 |
|
| [22] |
Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Chem. Phys. 2005, 311, 227.
doi: 10.1016/j.chemphys.2004.10.005 |
| [23] |
Stoychev, G. L.; Auer, A. A.; Neese, F. J. Chem. Theor. Comput. 2017, 13, 554.
doi: 10.1021/acs.jctc.6b01041 |
| [24] |
Peverati, R.; Truhlar, D. G. Philos. T. R. Soc. A 2014, 372, 20120476.
|
| [25] |
Werner, H.-J.; Knowles, P. J.; Manby, F. R.; Black, J. A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D. A.; Ma, Q.; Miller, T. F.; Mitrushchenkov, A.; Peterson, K. A.; Polyak, I.; Rauhut, G.; Sibaev, M. J. Chem. Phys. 2020, 152, 144107.
doi: 10.1063/5.0005081 |
| [26] |
Neese, F. WIREs Comput. Mol. Sci. 2012, 2, 73.
doi: 10.1002/wcms.81 |
| No related articles found! |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
