
A Theoretical Study of the Reaction of Fe+ with CH3X (X=Cl, Br, I)
Received date: 2013-02-05
Online published: 2013-03-26
Supported by
Project supported by the National Basic Research Program of China (973 Program) (No. 2012CB932800), the National Natural Science Foundation of China (Nos. 21103064, 21073075, 21173097), the Research Fund for the Doctoral Program of Higher Education of China (No. 20100061110046), the Special Funding of State Key Laboratory of Theoretical and Computational Chemistry, Jilin University and Basic Research Fund of Jilin University (Nos. 421010061439, 450060445067).
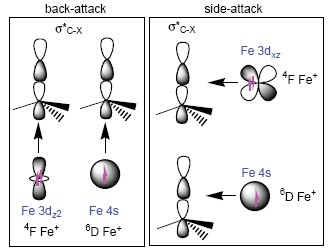
The C—X bond functionalization catalyzed by iron and their complexes has attracted much attention. Using density functional theory (DFT), we herein studied the reactivity and mechanism of iron cation (Fe+) towards C—X bond cleavage of CH3X (X=Cl, Br, I) at B3LYP/def2-SVP level of theory. The results show that there are two possible pathways available for the title reactions, i.e. the insertion mechanism and the SN2 mechanism, respectively. Mechanistically, in the insertion mechanism, the reactions stem from Fe+ attacking on the side of CH3X and results in the generation the products FeX+ and CH3·; whereas in the SN2 mechanism, the Fe+ initially attacks the substrate from the back of C—X yielding FeCH3+ and X·. The results show that the sextet and the quartet states of Fe+ demonstrate quite distinct reactivity towards the cleavage of C—X bonds in the most potential pathways, and the quartet pathways are dominant in all the pathways. The relative higher barriers in the SN2 pathways results in their lower competitiveness in the title reactions. In addition, our results show that, for all the three reaction systems, the insertion mechanism is exothermic; whereas for the SN2 mechanism, only X=I is exothermic, however. Furthermore, the calculations also show that these reactions demonstrate two-state reactivity (TSR) scenario. There are minimum energy crossing points (MECPs) between the sextet and quartet state on potential energy surfaces (PESs) both at the entrance and export sides for the two reaction mechanisms. On the other hand, the electron transfer evolution analysis indicates that the spin polarization plays important role in the stabilization of the potential energy surfaces and as a result, it controls the pathways by which it takes place and the branch ratio of the major products and byproducts. This thorough theoretical study, especially the detailed electronic structure analysis, may provide important clues for understanding the C—X bond activation and theoretical fundamental evidences for iron-based catalysts design.
Key words: Fe+; insertion; SN2; reaction mechanism; activation barrier; electronic structure
Sun Xiaoli , Li Jilai , Huang Xuri , Sun Chiachung . A Theoretical Study of the Reaction of Fe+ with CH3X (X=Cl, Br, I)[J]. Acta Chimica Sinica, 2013 , 71(05) : 749 -754 . DOI: 10.6023/A13020177
/
| 〈 |
|
〉 |
