


1 引言

2 结果与讨论
2.1 几何结构与稳定性


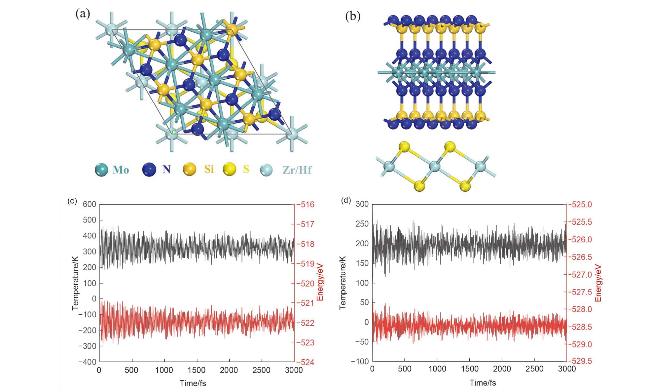
图3 (a) MoSi2N4, (d) ZrS2和(g) HfS2在300 K下的AIMD模拟结果, (b) MoSi2N4、(e) ZrS2和(h) HfS2的电子能带结构图及(c) MoSi2N4、(f) ZrS2和(i) HfS2的单层声子谱Figure 3 The AIMD simulation results of (a) MoSi2N4, (d) ZrS2, and (g) HfS2 at 300 K, electronic band structures of (b) MoSi2N4, (e) ZrS2 and (h) HfS2, and the single layer phonon spectra of (c) MoSi2N4, (f) ZrS2 and (i) HfS2 |
2.2 电子性质

图5 MoSi2N4/ZrS2 (a)和MoSi2N4/HfS2 (d)异质结的能带结构. MoSi2N4/ZrS2 (b)和MoSi2N4/HfS2 (e) vdW (van der Waals)异质结沿z方向的静电势图. MoSi2N4/ZrS2 (c)和MoSi2N4/HfS2 (f)异质结构的平均平面电荷密度图(蓝色和粉红色区域分别代表电子的积累和消耗)Figure 5 The band structure of MoSi2N4/ZrS2 (a) and MoSi2N4/HfS2 (d) heterojunctions. The Electrostatic potential diagrams of MoSi2N4/ZrS2 (b) and MoSi2N4/HfS2 (e) vdW heterojunctions along the z direction. The average plane charge density diagrams of MoSi2N4/ZrS2 (c) and MoSi2N4/HfS2 (f) heterojunctions (The blue and pink areas represent electron accumulation and consumption, respectively) |
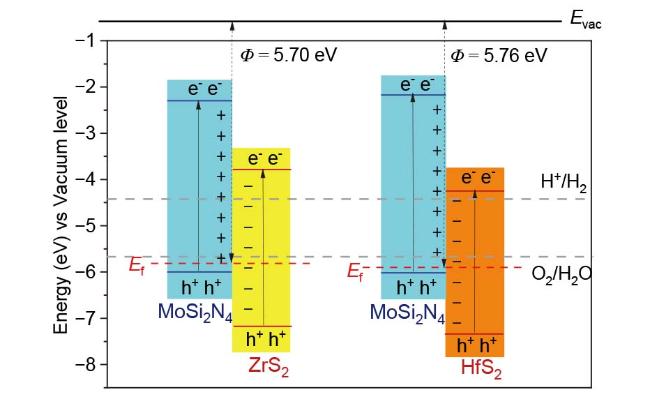
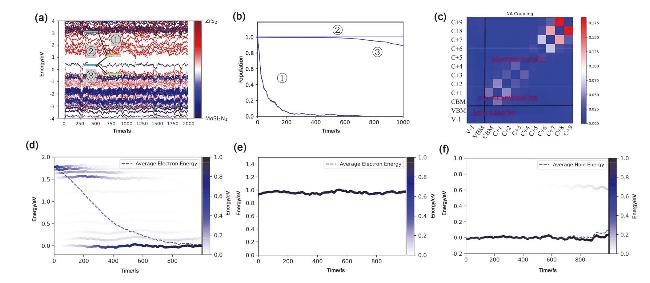
图7 MoSi2N4/ZrS2费米能级附近能量-时间变化图(a); 电子、空穴和电子-空穴复合途径载流子位置-时间图(b); 不同区域间非绝热耦合(NAC)的平均值图(c)(红色表示强耦合强度、蓝色表示弱耦合强度); (d)电子转移、(e)电子-空穴复合和(f)空穴转移能量随时间变化图(右侧颜色条表示不同能态的电子或空穴分布, 虚线表示平均电子或空穴能量)Figure 7 (a) Energy-time variation diagram near the Fermi level of MoSi2N4/ZrS2, (b) subcarrier position-time diagram of electrons, holes, and electron-hole recombination pathways, (c) averaged values of Nonadiabatic coupling (NAC) between different states. Time-dependent energy change for (d) electron transfer, (e) electron-hole recombination, and (f) hole transfer (The color bar on the right represents the distribution of electrons or holes in different energy states, and the dashed line represents the average electron or hole energy) |
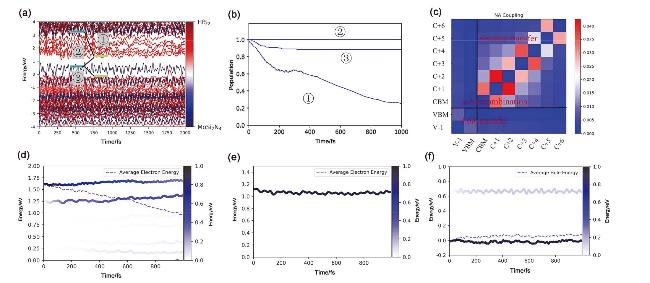
图8 MoSi2N4/HfS2费米能级附近能量-时间变化图(a); 电子、空穴和电子-空穴复合途径载流子位置-时间图(b); 不同区域间NAC的平均值图(c)(红色表示强耦合强度、蓝色表示弱耦合强度); (d)电子转移、(e)电子-空穴复合和(f)空穴转移能量随时间变化图(右侧颜色条表示不同能态的电子或空穴分布, 虚线表示平均电子或空穴能量)Figure 8 (a) Energy-time variation diagram near the Fermi level of MoSi2N4/HfS2, (b) Subcarrier position-time diagram of electrons, holes, and electron-hole recombination pathways. (c) Averaged values of NAC between different states. Time-dependent energy change for (d) electron transfer, (e) electron-hole recombination, and (f) hole transfer (The color bar on the right represents the distribution of electrons or holes in different energy states, and the dashed line represents the average electron or hole energy) |
表1 MoSi2N4/ZrS2(HfS2)异质结及其单层沿x、y方向的载流子迁移率Table 1 Carrier mobility of MoSi2N4/ZrS2(HfS2) heterojunction and its single layer along the x and y directions |
| 单层 | 方向 | 载波类型 | C/(N•m–1) | Ed/eV | m* (m0)a | μ/(cm2•V–1•s–1) | |
|---|---|---|---|---|---|---|---|
| ZrS2 | x | e | 5.191 | –1.29 | 1.971 | 178.785 | |
| h | –4.94 | –1.719 | 16.119 | ||||
| y | e | 6.788 | –1.95 | 0.657 | 927.459 | ||
| h | –4.83 | –0.573 | 198.751 | ||||
| MoSi2N4 | x | e | 38.788 | –16.57 | 2.236 | 6.342 | |
| h | –6.59 | –4.917 | 8.303 | ||||
| y | e | 38.964 | –12.62 | 0.745 | 98.861 | ||
| h | –6.54 | –1.639 | 75.998 | ||||
| HfS2 | x | e | 7.072 | –1.59 | 1.855 | 181.751 | |
| h | –5.17 | –1.767 | 19.022 | ||||
| y | e | 7.302 | –1.58 | 0.618 | 1701.824 | ||
| h | –5.06 | –0.589 | 184.695 | ||||
| MoSi2N4/ ZrS2 | x | e | 50.568 | –4.58 | 0.698 | 1112.332 | |
| h | –8.43 | –0.423 | 890.465 | ||||
| y | e | 50.807 | –4.58 | 0.233 | 10029.576 | ||
| h | –8.85 | –0.141 | 7299.525 | ||||
| MoSi2N4/ HfS2 | x | e | 52.350 | –4.54 | 0.689 | 1200.605 | |
| h | –5.35 | –1.340 | 228.467 | ||||
| y | e | 52.556 | –4.52 | 0.229 | 11027.355 | ||
| h | –5.36 | –0.447 | 2058.138 |
a m*是基于m0进行计算得到的,m0是指电子的静止质量, 其数值约为9.11×10–31 kg. |
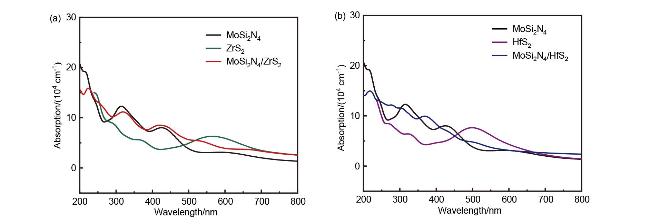
2.3 光吸收
2.4 催化性能
表2 MoSi2N4和MoSi2N4/ZrS2(HfS2)的产氢效率Table 2 ηSTH of the MoSi2N4 and MoSi2N4/ZrS2(HfS2) |
| Structure | χ(H2)/eV | χ(O2)/eV | Eg/eV (HSE) | ηabs/% | ηcu/% | ηSTH/% |
|---|---|---|---|---|---|---|
| MoSi2N4 | 0.96 | 0.08 | 2.27 | 25.58 | 15.15 | 3.88 |
| MoSi2N4/ZrS2 | 0.81 | 1.01 | 1.39 | 67.73 | 60.44 | 40.94 |
| MoSi2N4/HfS2 | 0.85 | 1.08 | 1.66 | 53.08 | 54.85 | 29.12 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}