

1 引言
环金属Ir(III)配合物因其可调的光物理性质和化学稳定性而在光电子领域具有广泛的应用. 许多环金属Ir(III)配合物因其高的发光量子产率和大的Stokes位移而成为有机发光二极管(OLED)和有机太阳能电池(OSC)技术中的高效发射体, 能够发射出具有优异色纯度和长寿命的绿光、红光或蓝光[1-3]. 某些Ir(III)配合物显示出长余辉效应, 可用于显示和安全标记[4-5]. 环金属Ir(III)配合物还显示出较高的细胞摄取效率, 定位于特定的亚细胞器, 在生物组织中具有特异性分布, 可作为荧光标记物而用于生物成像领域[6-9], 不少配合物还被用作细胞内信号分子(如H2S、O2和H2O2等)的荧光探针[10-11]. 环金属Ir(III)配合物表现出较低的暗细胞毒性和较高的光毒性, 甚至有一些Ir(III)配合物还对顺铂耐药肿瘤细胞表现出良好的毒活性. 由于其独特的抗肿瘤特性, 环金属Ir(III)配合物已作为光敏剂用于光动力治疗, 它们可以在光激发下产生活性氧物种(ROS)并靶向癌细胞[12-15], 导致细胞坏死或凋亡.
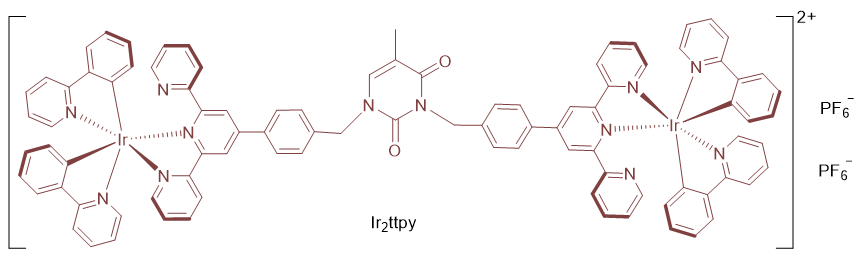
大多数环金属Ir(III)配合物采用{N,C}配位模式, 常用的{N,C}配体是基于苯基吡啶或苯并喹啉[7,16]的衍生物. 由于Ir(III)核的饱和配位数为6, 因此通常使用联吡啶、三联吡啶或邻菲咯啉衍生物作为辅助配体. 鉴于2,2':6',2''-三联吡啶的优异配位能力, 且大多数三联吡啶金属配合物表现出独特的光学性质[17], 考虑到胸腺嘧啶拥有两个便于修饰的酰胺基团, 将其与2,2':6',2''-三联吡啶基团连接起来, 制备多联吡啶衍生物ttpy(图1). 胸腺嘧啶的引入, 有可能提升配体分子的生物相容性与靶向性[18-19]. 选择苯基吡啶作为共同配体, 进而制备出含有正二价配位阳离子的双核铱配合物Ir2ttpy(图1). 本工作进一步研究了ttpy和Ir2ttpy的光学活性和抗肿瘤活性.
2 结果与讨论
2.1 合成与结构表征
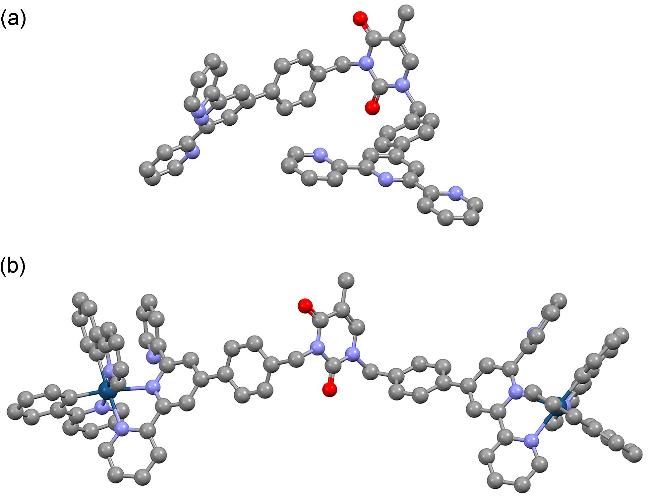
胸腺嘧啶分子中两个O=CNH基团的酸性不同, 理论上可以采用不同强碱(如KOH和t-Bu-OK)进行分步脱氢, 再与4'-p-溴亚甲基苯基-2,2:6',2''-三联吡啶进行1∶1或1∶2亲核取代反应, 从而可控地在胸腺嘧啶分子上连接一个或两个三联吡啶基团. 由于分步脱氢再进行1∶1取代反应的产物的分离纯化困难, 本工作采用NaH进行完全脱氢, 然后进行1∶2取代反应得到目标配体ttpy. 需要注意的是, NaH不可过量. Ir2(bpy)4Cl2中的Ir—Cl桥键稳定性较差, 可以在温和的条件下被三联吡啶基团中的N原子取代配位, 形成更稳定的Ir—N 键[20], 电喷雾质谱和核磁氢谱的数据表明得到的是双核铱配合物.
2.2 配体及铱配合物的单光子吸收与发射性能
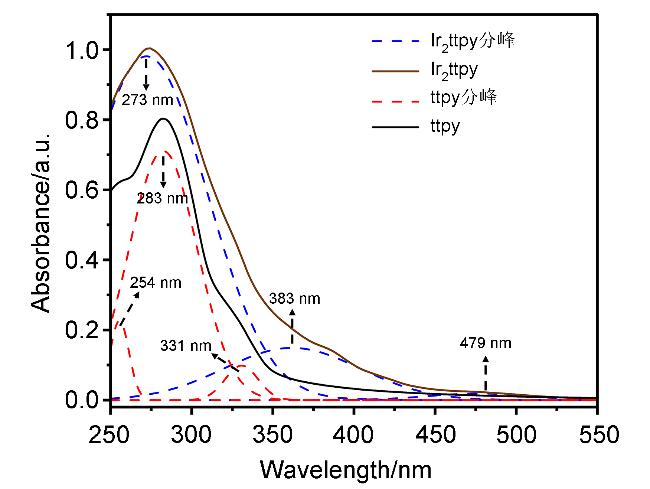
胸腺嘧啶基团在紫外区的最大吸收峰位于254 nm. 当其与两个4'-苯基-2,2':6',2''-三联吡啶基团连接后, 产生的强烈ILCT吸收峰位于283 nm(图3). 与大部分的三联吡啶衍生物类似, ttpy中2,2':6',2''-三联吡啶基团的π→π*吸收峰具有中等强度, 在UV-Vis吸收谱中呈现出肩峰, 最大吸收波长位于331 nm. 配合物Ir2ttpy中源自胸腺嘧啶基团的吸收波长相较于配体的蓝移10 nm, 吸收峰的强度显著增加, 摩尔吸收系数为9.8×104 L• mol-1•cm-1. 三联吡啶基团采取{N,N}双齿配位, 其π→π*吸收峰相较于配体显著红移, 最大吸收波长位于383 nm. Ir2ttpy中的配位单元与[Ir(ppy)2(bpy)]极为相似, 因此其3MLCT强度也较弱. 与此同时, 来源于tpy配体以及嘌呤连接基团的1LC跃迁吸收峰相较bpy的更强, 使得3MLCT在吸收谱中(400~550 nm范围内)表现为几不可见的坡峰, 峰值位于479 nm. 这一现象在其他含有嘌呤基团的铱配合物中也有呈现[25-27].
为了进一步明确配体与配合物吸收峰的跃迁机制, 采用含时密度泛函(TDDFT)计算其基态和激发态的结构、能级以及电子云分布(图4). 配体ttpy中ILCT吸收峰(HOMO-1→LUMO+2, E=4.43 eV)是电子云从胸腺嘧啶基团转移到三联吡啶部分, 而π→π*吸收峰(HOMO→LUMO, E=3.99 eV)是电子云从三个吡啶环蔓延到相邻的苯环上. 在双核配合物Ir2ttpy中, 由于2-苯基吡啶的贡献, 其π→π*吸收峰(HOMO-8→LUMO+1, E=3.44 eV)与配体ttpy的相比显著红移了50 nm, 与实验测试结果一致. MLCT吸收峰(HOMO→LUMO, E=2.58 eV)的强度较弱. 虽然配体与配合物分子中都连接有两个三联吡啶基团, 但ILCT、π→π*、MLCT的吸收峰都只有一个三联吡啶基团的电子云参与其中.
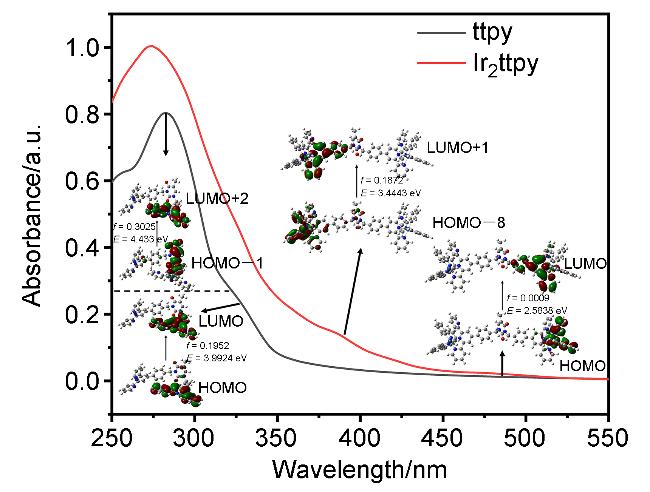
图4 配体ttpy与配合物Ir2ttpy的紫外-可见吸收光谱指认结果(通过TD-DFT计算得到ttpy和Ir2ttpy优化几何结构的分子轨道. 使用TD-B3LYP方法和6-31G(d)基组计算跃迁能, 单位为eV)Figure 4 Assignment for the UV-vis spectra of ligand ttpy and complex Ir2ttpy (Molecular orbitals for ttpy and Ir2ttpy were optimized from TD-DFT calculations. Transition energies were calculated using the TD-B3LYP method with 6-31G(d) basis sets, eV) |
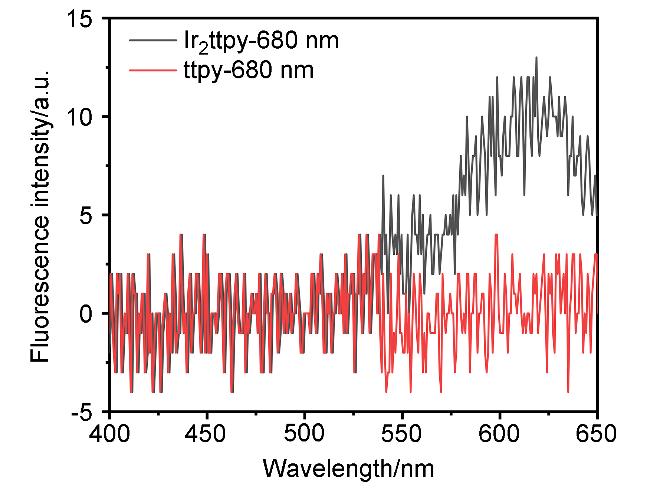
配体ttpy中, 两个4'-苯基-2,2':6',2''-三联吡啶基团与胸腺嘧啶仅通过单键相连, 没有生成更大共轭体系, 因此仅表现出基于三联吡啶端的3LC跃迁的荧光发射峰, 峰值位于350 nm. 配合物Ir2ttpy荧光发射谱呈现两个发射峰, 350 nm的峰相较于配体的明显减弱, 在600 nm处呈现出基于3MLCT的宽峰. 3LC与3MLCT峰值差达到250 nm(图5), 这种双重发射性能在荧光探针以及细胞成像领域有着一定的应用价值[28]. 溶剂的极性对配体ttpy与配合物Ir2ttpy的荧光发射波长几乎没有影响(支持信息图S3和S4), 仅影响到发射峰的强度, 可能是由于分子结构具有强稳定性, 致使其激发和发射过程并不依赖于溶剂的极性. 常见环金属铱配合物的发光寿命可达μs级, 而Ir2ttpy的荧光寿命仅为105 ns, 可能由于胸腺嘧啶与三联吡啶基团之间自由旋转的C—C单键可通过非辐射跃迁的方式散失激发态能, 缩短了激发态寿命. Ir2ttpy量子产率小于1%(支持信息图S5和S6), 相较文献报道的不少铱配合物的结果偏低[1,7].

2.3 配体及铱配合物的双光子吸收与发射性能
文献中报道了大量的多联吡啶衍生物及其环金属铱配合物, 他们表现出一定的双光子吸收(TPA)与双光子发射(TPEF)性能[29-30]. 配体ttpy中胸腺嘧啶与三联吡啶未能形成更大的共轭体系, 且分子的整体平面性较差, 在650~1000 nm范围内没有检测到TPA信号. 与配体ttpy不同, 配合物Ir2ttpy在650~800 nm范围内表现出明显的双光子吸收效应. 开孔Z扫描拟合结果显示, 配合物Ir2ttpy在680 nm激光照射下显示出最强的TPA信号, 其双光子截面为24 GM(图6). 双光子吸收可归属于三联吡啶基团的π→π*跃迁, 四个苯基吡啶基团可能也有一定的贡献. 与单光子吸收谱中MLCT吸收峰信号很弱的现象一致, 配合物Ir2ttpy在800~1100 nm范围内没有观测到TPA信号. 配合物Ir2ttpy在650~1100 nm的飞秒激光的激发下, 产生一定的双光子荧光发射性能. TPEF发射只呈现出一个宽的单峰, 在500~700 nm范围内的峰形与单光子荧光发射峰接近, 且峰值位于600 nm左右, 表明TPEF也源自3MLCT激发态. 配体ttpy没有检测到TPEF信号(图7).
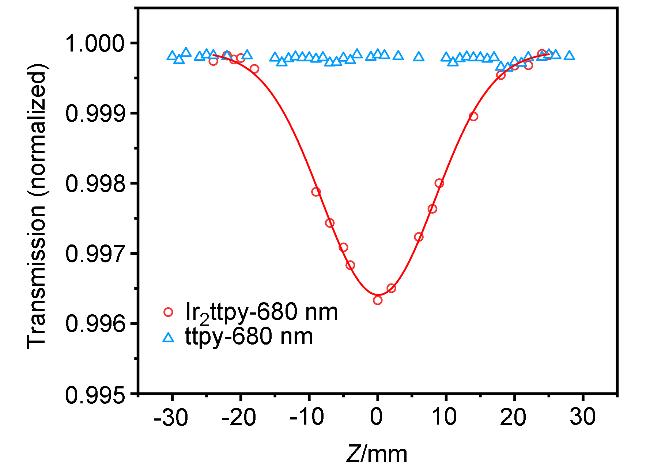
图6 配体ttpy与配合物Ir2ttpy在680 nm飞秒激光激发下的开孔Z-扫描结果及高斯拟合曲线(实线)Figure 6 Open-aperture Z-scan experimental data and fitting curve for complex Ir2ttpy under a 680 nm femtosecond laser beam |
2.4 铱配合物的抗肿瘤活性
用噻唑蓝(MTT)法来评测铱配合物Ir2ttpy的体外细胞毒活性. 选取鼠源乳腺癌细胞(4T1)为研究对象, 经过48 h孵化, 即使配合物的浓度已达125 μmol/L, 4T1细胞的存活率仍高于60%, 表明配合物的暗毒性较差, IC50约为237 μmol/L.
作为对照, 采用450 nm的光照射30 min(功率30 mW•cm−2), 配合物的细胞毒活性明显升高(支持信息图S7), IC50约为13 μmol/L, PI=18.
选用人正常干细胞系(LO2)细胞进行光毒性和暗毒性测试, 发现Ir2ttpy也能对LO2造成一定的损伤, 但明显比对癌细胞的生长抑制活性低.
2.5 铱配合物的细胞内定位
文献中不少环金属铱配合物表现出一定的抗肿瘤活性, 研究表明这些铱配合物可与多种细胞器如线粒体、溶酶体、高尔基体、内质网和脂滴等发生相互作用. 如对线粒体的形态进行实时监控可以发现, 配合物能诱导线粒体损伤从而杀死癌细胞[15]. 近年来线粒体选择性定位的铱配合物备受关注, 研究者们认为光诱导的细胞内氧化还原失衡及线粒体膜电位的改变等导致了肿瘤细胞的坏死和凋亡损伤.
根据文献中铱配合物[Ir(ppy)2(N^N)]X的抗肿瘤机理的研究结果(定位线粒体/溶酶体且导致线粒体/溶酶体功能损伤), Ir2ttpy因其配位单元与之结构类似, 预期其也有可能进入线粒体或溶酶体. 将之与线粒体染色剂MitoTracker Deep Red (MTDR)和溶酶体染色剂Cell Lyso Tracker Red (LTR)对Hela细胞进行共染, 通过共聚焦显微成像初步分析其靶向线粒体或溶酶体的可能性.
由于Ir2ttpy本身的荧光不强, 其在活细胞环境下还会发生荧光部分淬灭, 即使将配合物Ir2ttpy的给药浓度提高到50 μmol/L, 进入细胞的Ir2ttpy的荧光发射强度还是非常弱, Merge组中无明显重叠荧光信号(支持信息图S8). 经过多次尝试(根据Ir2ttpy的IC50数据, 调整共染实验的给药浓度), 目前未能确证Ir2ttpy能被线粒体或溶酶体有特异性的摄入.
2.6 配体及铱配合物的DNA结合性能
鉴于DNA是Pt(II)、Ru(II)等金属抗癌药物的可能靶分子, 铱配合物也可能会与细胞核内DNA或者线粒体DNA发生一定的相互作用, 从而影响DNA的分子结构和生物功能[31].
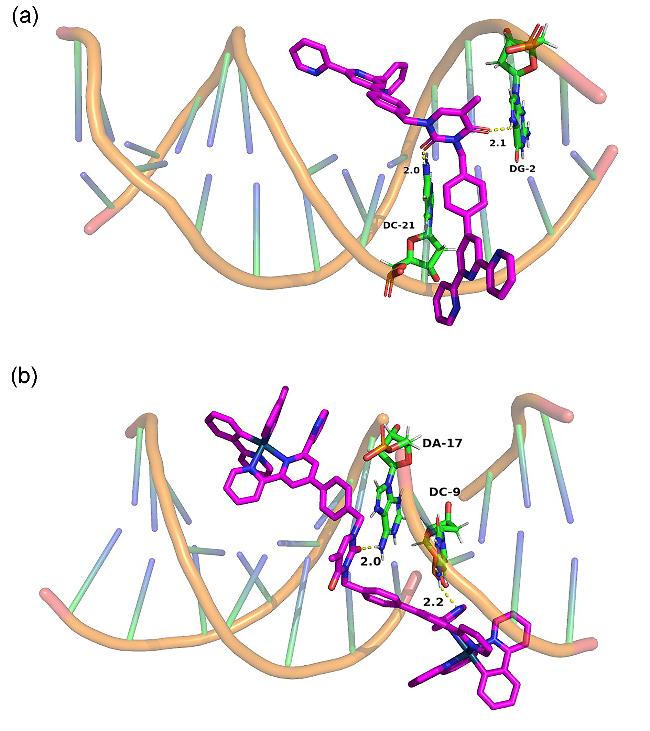
由图8可见, 随着CT-DNA浓度逐渐增加, 配体和配合物的紫外-可见吸收光谱表现出明显的减色效应. ttpy与Ir2ttpy分别在304和334 nm处出现一个明显的等吸收点, 表明它们都能与DNA发生一定的相互作用, 且能形成较为稳定的复合物. 根据等吸收点出现的位置, 推测ttpy中的胸腺嘧啶基团或者三联吡啶端与DNA可能形成氢键, 而Ir2ttpy的配位中心能通过氢键或者通过静电结合形成复合物.

图8 ttpy (a)和Ir2ttpy (b)的紫外可见吸收光谱随DNA浓度递加的变化图(化合物的浓度为2.0×10-5 mol/L, 缓冲溶液组成: 5 mmol/L tris, 50 mmol/L NaCl, pH=7.42, r=[CT-DNA]/[ttpy or Ir2ttpy]=0~1.0.)Figure 8 UV absorption spectra of ttpy (a) and Ir2ttpy (b) solution with increasing concentration of CT-DNA [buffer=5 mmol/L tris, 50 mmol/L NaCl, pH=7.42. The concentration of ttpy (or Ir2ttpy) is 2.0×10-5 mol/L, r=[CT-DNA]/[ttpy/or Ir2ttpy]=0~1.0] |
2.7 配体及铱配合物的ROS生成性能
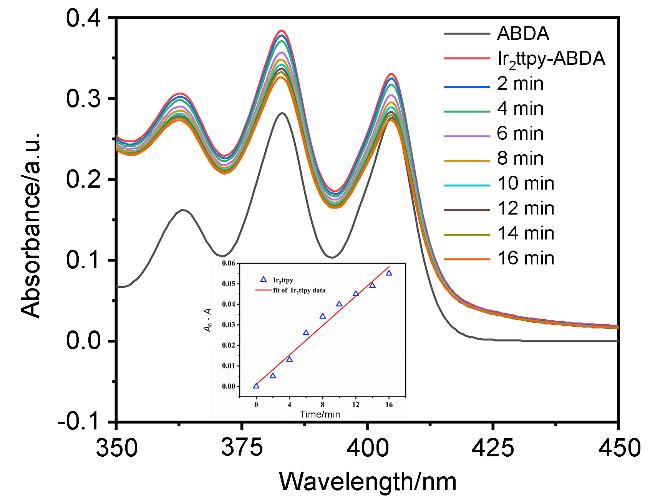
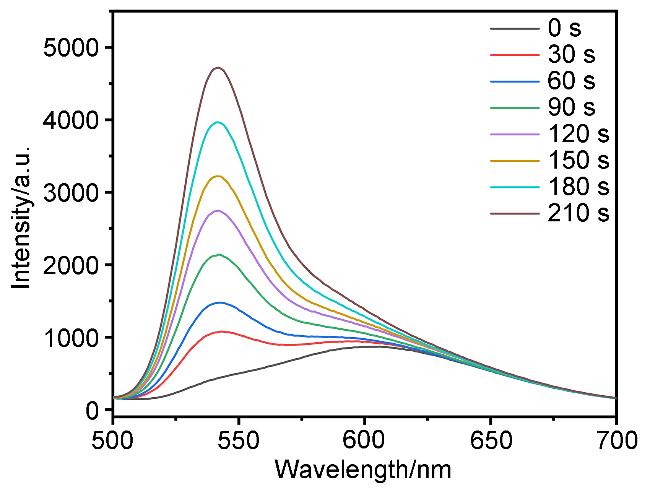
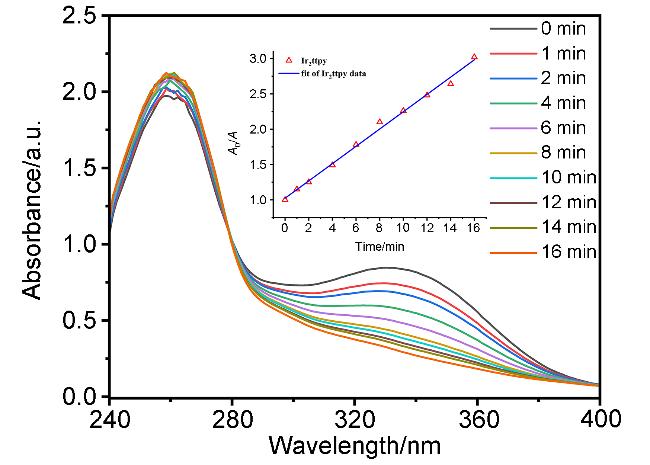
烟酰胺腺嘌呤二核苷酸(NADH)作为一种还原辅酶在生物催化过程中充当着重要角色. 在线粒体中, NADH是电子传递链的重要供体, 其携带的电子参与氧化磷酸化过程以产生三磷酸腺苷(ATP). ROS可以氧化NADH, 使其转化为NAD⁺. 这种氧化作用会影响NADH/NAD⁺的比例, 进而影响细胞的氧化还原状态和能量代谢途径[37]. 鉴于配合物Ir2ttpy能在光诱导下生成ROS, 采用紫外可见吸收光谱跟踪其催化氧化NADH的反应. 从图12可以看出, NADH在340 nm处的吸光度明显减弱, 表明配合物Ir2ttpy能在可见光照射下有效地将NADH转化为NAD+. 由340 nm处的吸光度差值可算出Ir2ttpy与NADH之间催化反应的转化数(TON)为22.4, 转换频率(TOF)为89.2 h-1, 光照16 min后NADH吸收峰几乎完全消失.
采用CellROX™来监测细胞内ROS的生成情况. CellRox™绿色试剂是一种用于ROS的荧光开启型指示剂[38]. 当HeLa细胞仅与Ir2ttpy或CellROX™绿色试剂孵育时, 未检测到荧光信号(图13中的第1组和第2组). 当在细胞培养物中同时加入Ir2ttpy和CellROX™后, 出现了可测量的荧光发射, 这表明即使在黑暗条件下, 存在Ir2ttpy时细胞内ROS的水平也会升高(图13中的第3组). 值得注意的是, 短时间(1 min)光照显著增强了荧光, 这一结果是由于新产生的细胞内ROS对CellROX™的氧化作用(下图中的第5组). 此外, 使用经N-乙酰-L-半胱氨酸(NAC, 一种抗氧化剂)预处理的细胞, 产生了淬灭的荧光发射(图13中的第4组和第6组).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.8 配合物抗肿瘤的可能机理
在细胞环境中, 铱配合物光诱导产生的ROS以及与DNA的相互作用共同影响细胞命运. 铱配合物在光激发下产生的ROS具有高度活性, 能够导致碱基氧化、糖基损伤以及DNA链断裂等多种形式的损伤, 严重影响DNA的完整性和稳定性. ROS造成的DNA损伤若超过细胞自身的修复能力, 会触发细胞周期阻滞、凋亡等一系列细胞反应[39].
铱配合物与DNA的结合可能进一步干扰DNA相关的生物过程, 如转录和复制, 加剧细胞毒性, 协同诱导细胞凋亡. 铱配合物与DNA的结合会影响配合物吸收光能的效率以及激发态的寿命. 这种结合可能改变配合物周围的微环境, 影响能量传递过程, 从而调节ROS的生成量, 也会影响ROS的生成路径和种类.
目前对于铱配合物光诱导产生ROS以及其与DNA相互作用的研究, 在检测技术上存在一定局限性, 难以实时、原位地精确监测ROS的生成动态以及铱配合物在DNA上的结合位点变化.
3 结论
制备了基于胸腺嘧啶三联吡啶配体的双核环金属铱配合物Ir2ttpy, 其单光子吸收谱中呈现出强ILCT和弱MLCT吸收峰, 单光子发射谱中的双重荧光发射峰分别来自3LC和3MLCT激发态, 激发态寿命约为101 ns, 量子产率小于1%. 最大双光子吸收截面约为24 GM, 双光子激发发射峰位于600 nm, 源自3MLCT激发态. 配合物Ir2ttpy针对4Tl乳腺癌细胞表现出一定的光毒性, PI值达到18. 配合物Ir2ttpy可以嵌入DNA大沟区, 以氢键和静电吸引力与DNA发生相互结合作用. 配合物Ir2ttpy在可见光照射下能够生成单线态氧和超氧负离子, 对NADH有明显的催化氧化效应. 配合物Ir2ttpy的抗肿瘤活性与其诱导生成ROS以及与DNA相互结合有关, 二者协同作用而导致肿瘤细胞死亡.
4 实验部分
4.1 仪器与设备
未特别指明的化学品都来源于商业采购并直接使用. CT-DNA从Sigma-Aldrich公司购买. 所有合成的化合物均在真空干燥器(CaSO4)中干燥后再进行结构表征. 1H NMR用500 MHz Bruker DMX分析, TMS作内标. ESI-MS数据用Agilent 1290-6545 LC/MS采集, 元素分析数据用Heraeus CHN-O快速分析仪采集. 紫外-可见吸收光谱用PerkinElmer LAMBDA®365记录, 荧光光谱用Hitachi F-7000记录. 荧光寿命和量子产率采用HORIBA FLUOROLOG-3-11进行测量. CT-DNA的浓度用紫外光谱标定(ε260nm=6600 L•mol-1•cm-1). 在Tris- HCl的缓冲溶液中(pH=7.42), 260和280 nm处紫外吸光度的比值为1.87, 表明DNA中已完全去除了蛋白质杂质.
4.2 化合物的合成与表征
4.2.1 配体ttpy的合成
将0.126 g胸腺嘧啶(1 mmol)溶于8 mL无水N,N-二甲基甲酰胺(DMF)后, 通入N2同时将溶液升温至40 ℃, 加入0.048 g NaH (2 mmol), 充分搅拌反应1 h. 将0.804 g (2 mmol) 4'-p-溴亚甲基苯基-2,2':6',2''-三联吡啶溶于20 mL DMF后, 再逐滴将其加入到上述溶液中, 在氮气保护下反应24 h. 减压蒸除溶剂, 得到黄白色固体. 再用100 mL乙醇进行重结晶处理, 得到白色固体256 mg, 产率27.5%. 1H NMR (DMSO-d6, 500 MHz) δ: 8.78 (d, J=4.0 Hz, 4H), 8.69 (q, J=8.0 Hz, 8H), 8.04 (t, J=7.5 Hz, 4H), 7.97~7.95 (m, 3H), 7.91 (d, J=8.5 Hz, 2H), 7.57~7.51 (m, 8H), 5.15 (s, 2H), 5.07 (s, 2H), 1.90 (s, 3H); HRMS (ESI) calcd for C49H36N8O2Na 791.2853, found 791.2855.
4.2.2 配合物Ir2ttpy的合成
将100 mg (0.09 mmol) Ir2(bpy)4Cl2和60 mg (0.08 mmol) ttpy溶于30 mL二氯甲烷和甲醇混合溶液(V∶ V=1∶1)中, 在65 ℃下加热回流24 h, 全程N2保护且避光处理. 反应结束后减压蒸除溶剂, 得到橙色固体. 将橙色固体溶于50 mL水与DMSO的混合溶液(V∶V=4∶1)中, 加入150 mg NH4PF6固体, 继续搅拌30 min, 过滤收集橙色沉淀物, 用水(20 mL×3)、乙醇(10×3 mL)和乙醚(10 mL)洗涤, 真空干燥后得橙色粉末92 mg, 产率57.5%. 1H NMR (acetone-d6, 500 MHz) δ: 9.12 (t, J=8.0 Hz, 6H), 8.82 (s, 1H), 8.77 (q, J=8.0 Hz, 2H), 8.33 (t, J=8.5 Hz, 2H), 8.28~8.25 (m, 2H), 8.20 (t, J=8.0 Hz, 2H), 8.11 (d, J=8.0 Hz, 4H), 8.01 (t, J=8.0 Hz, 4H), 7.91 (t, J=8.0 Hz, 4H), 7.81 (d, J=7.5 Hz, 2H), 7.76 (d, J=6.0 Hz, 2H), 7.68~7.62 (m, 6H), 7.50 (d, J=8.0 Hz, 2H), 7.32 (t, J=7.5 Hz, 2H), 7.26 (t, J=7.5 Hz, 2H), 7.08 (t, J=6.0 Hz, 2H), 7.04 (t, J=6.0 Hz, 2H), 6.95 (t, J=7.5 Hz, 2H), 6.87 (d, J=7.5 Hz, 2H), 6.76 (t, J=7.5 Hz, 2H), 6.60 (t, J=7.5 Hz, 2H), 6.31 (t, J=7.5 Hz, 2H), 5.96 (d, J=7.5 Hz, 2H), 5.53 (d, J=7.5 Hz, 2H), 5.21 (d, J=14.5 Hz, 2H), 5.14 (d, J=14.5 Hz, 2H), 1.90 (3, 3H); HRMS (ESI) m/z calcd for C93H68N12O2Ir22+ 885.0415, found 885.2489.
4.3 化合物的双光子性能研究
在开孔Z扫描装置上测定双光子吸收截面δ2. 所有的光学测试都是使用脉冲持续时间为140 fs、重复频率为80 MHz的飞秒激光进行的. 使用1 kHz机械斩波器消除激光脉冲对Ir(III)配合物的加热效应. 在开孔条件下评估非线性吸收, 根据以下方程对测量的实验数据进行拟合.
$T(z)\frac{1}{q(z)\sqrt{\text{ }\!\!\pi\!\!\text{ }}}\int_{\infty }^{\infty }{\ln [1q(z)}]\exp ({{\tau }^{2}})\text{d}\tau $
双光子激发发射光谱利用锁模钛宝石激光器(Coherent Mira900F, 200 fs, 76 MHz)作为激发光源, 采用单扫描条纹相机(Hamamastu C5680-01)来进行光谱记录. 样品溶于H2O/DMSO (V∶V=1∶1)混合溶液, 浓度1.0×10−3 mol•L−1.
4.4 密度泛函理论(DFT)计算
配体和配合物的DFT计算采用Gaussian 09程序包进行, 利用B3LYP密度泛函理论对分子结构进行了优化. 在基态和激发态优化的基础上, 应用含时密度泛函(TDDFT)方法研究了激发态的电子性质. 在计算过程中, 配体的所有原子采用6-31g(d)基组, 配合物采用了6-31g(d)(非金属原子)/LANL2DZ (Ir原子)混合基组. 在结构优化过程中, 采用了Gaussian 09程序的默认收敛标准, 即能量收敛标准为1.0×10-6 Hartree, 最大力收敛标准为3.0×10-4 Hartree/Boh, 均方根力收敛标准为1.0×10-4 Hartree/Bohr, 最大位移收敛标准为1.2×10-3 Bohr, 均方根位移收敛标准为8.0×10-4 Bohr.
4.5 与DNA相互作用的分子模拟
采用Autodock程序对配合物与DNA之间的相互结合进行模拟. Ir(III)配合物的结构通过DFT (B3LYP)方法优化得到, DNA (PDB ID 1BNA)的分子结构从蛋白质数据库中获得(http://www.rcsb.org/pdb). 在建模时, DNA- Ir2ttpy的网格盒大小为6.8 nm×7.6 nm×12.6 nm, 网格间距为0.0375 nm. 运用拉马克遗传算法(LGA)计算结合在DNA上的Ir2ttpy可能出现的构象, 进行100次GA运算, 其他参数使用默认值. 利用Pymol软件分析结合自由能最低的构象的对接结果.
(Zhao, C.)