


1 引言
碳酸二甲酯(DMC)是一种绿色环保的精细化学品.由于DMC本身具有极性, 粘度低, 且较为稳定, 可作为优良有机溶剂[1-2]. DMC可以代替甲基叔丁基醚作为绿色环保的汽油添加剂用于降低油品的凝固点, 调控其辛烷分布以及提升抗爆性[3-4]. 由于具有较高的离子导电性, DMC在锂电池行业也被广泛用作电解液[5-6]. DMC的生产技术包括光气法[7]、酯交换法[8]、尿素醇解法[9-10]、CO2直接合成法[11]和CO酯化法. 其中CO酯化法在DMC合成过程中不产生水, 有利于稳定催化剂, 且反应条件温和, 具有独特的技术优势, 因此受到了广泛关注. 目前已经报道了一些含氯催化剂[12-14]和无氯催化剂[15-16], 尽管含氯催化剂表现出较高的初始催化活性和选择性, 但由于反应过程中氯离子会以氯甲酸甲酯副产物的形式不断流失, 导致催化剂迅速失活, 为了维持含氯催化剂的稳定性, 需要不断通入干燥的氯化氢气体, 造成了设备严重腐蚀, 增加了生产成本. 相比之下, 无氯催化剂可以摆脱氯离子的限制. 郭国聪等[17]开发了一种孤立态的Pd(Ⅱ)/NaY无氯催化剂, 该催化剂能在100 h内保持对DMC的超高选择性(>99.5%). 报道的无氯催化剂的载体通常为NaY和UiO-66[18-22]. 然而, NaY的酸性会造成原料亚硝酸甲酯(MN)的部分分解[23], 产生不需要的副产物, 因此研发非分子筛体系的催化剂具有重要意义.
由于CeO2酸性较弱, 原料MN分解问题几乎不存在, 然而Pd/CeO2催化剂的DMC选择性却较低. 为了提升DMC选择性, 我们合成了不同Pr掺杂比例(Pr与CeO2的物质的量比2.5%, 5%, 7.5%, 10%)的Pd/Pr-CeO2催化剂, 并评价了它们在CO酯化生成DMC中的反应性能. 结果发现, 催化剂的DMC选择性与其氧空位浓度成正比. X-射线光电子能谱(XPS)和氢气程序升温还原(H2-TPR)揭示了不同催化剂中Pd物种的电子结构以及金属-载体相互作用的差异. 氧空位能增强金属与载体之间的相互作用, 有利于保持Pd(Ⅱ)高度分散而不易被反应气中的CO还原, 从而有利于选择性生成DMC.
2 结果与讨论
2.1 催化性能评价
首先对四种Pr-CeO2载体进行了MN分解测试, 结果如支持信息表S1所示. 与NaY分子筛相比, MN在Pr-CeO2载体上的分解少很多, 这是因为NaY酸性较强[23], 而CeO2偏弱碱性. 将四种催化剂在相同反应条件下用于CO酯化制DMC反应中, 它们的CO转化率和DMC选择性分别如支持信息图S1和图1所示. Pd/Pr-CeO2-2.5和Pd/Pr-CeO2-10表现出较好的催化活性, CO转化率分别为59%和56%, 略高于Pd/Pr-CeO2-5和Pd/Pr-CeO2-7.5的CO转化率(44%). 但Pd/Pr-CeO2-5具有最高的DMC选择性(77%), 远高于其它三种催化剂. 四种催化剂的制备方法相同, 仅载体掺杂Pr的比例不同, 为了揭示催化剂存在显著催化选择性差异的原因, 我们进行了以下一系列表征.
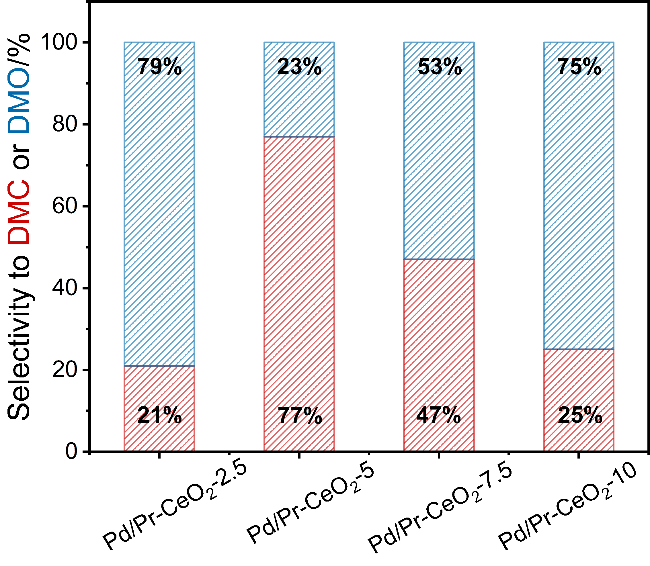
图1 四种催化剂在CO酯化制DMC反应中DMC和DMO的选择性 (反应条件: 200 mg催化剂, 0.1 MPa, 120 ℃, 空速为2500 L•kgcat-1•h-1, V(CH3ONO)/V(CO)=2.4, 反应时间6 h)Figure 1 The selectivities of DMC and DMO in the esterification of CO to DMC reaction with four catalysts (Reaction conditions: 200 mg of catalysts, 0.1 MPa, 120 ℃, space velocity of 2500 L•kgcat-1•h-1, V(CH3ONO)/V(CO)=2.4, reaction for 6 h) |
2.2 催化剂结构表征与分析
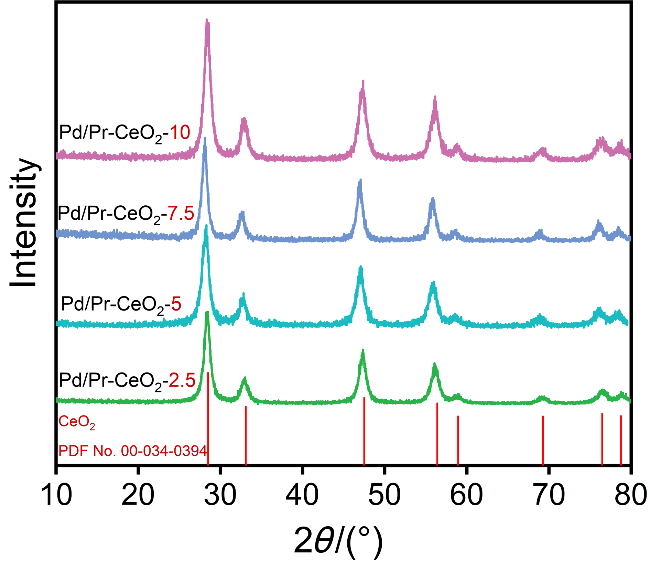
支持信息图S2和图2分别是合成的载体和催化剂的X射线衍射(XRD)图谱. 合成的不同样品均在28.5°、33.1°、47.5°、56.3°、59.1°、69.4°、76.7°和79.0°(PDF No. 00-034-0394)处出现衍射峰, 而这些衍射峰的位置分别对应的是CeO2萤石型立方结构(JCPDS 89-8436)的平面衍射图谱的各晶面, 确认成功合成了Pr-CeO2载体. 且在图谱中未观察到Pr2O3对应的衍射峰, 说明所合成的载体中Pr的掺杂作用并不是形成Pr2O3, 而是成功掺杂在CeO2晶格中. Pr-CeO2载体晶体结构示意图如支持信息图S3所示.在不同的Pd/Pr-CeO2催化剂的XRD图谱中都没有观察到Pd的衍射峰(39.6°), 说明活性组分Pd的尺寸较小且在载体表面分散较好. 各催化剂的晶格常数列于支持信息表S2, 所有催化剂的晶格常数均大于Pd/CeO2, 进一步证实了Pr成功掺入CeO2载体中. 支持信息图S4是各催化剂反应后的XRD图谱, 与反应前相比, 各个衍射峰的位置和强度没有发生明显变化.
合成的催化剂的透射电子显微镜(TEM)和高分辨透射电子显微镜(HRTEM)图像如图3所示, 在低倍电镜下可以观察到所有制备的样品均为棒状结构, 这些纳米棒的长度约为30~150 nm, 宽度约为8~15 nm, 说明棒状载体制备较为成功. 四种Pd/Pr-CeO2催化剂中的晶格间距均约为0.335 nm, 对应的是(111)晶面且未发现有其他晶面的存在. 与纯CeO2纳米棒的晶格间距d111=0.312 nm相比, Pr掺杂CeO2纳米棒的晶格常数表现出晶格膨胀[29], 这可能是因为Pr3+离子(0.113 nm)进入CeO2的晶格, 取代了Ce4+离子, 导致了晶格畸变的发生[30]. 此外, 在纳米棒表面存在大量的缺陷, 在图上显示为棒体上的白色斑点以及边缘的不规则锯齿状的缺陷. Pd/Pr-CeO2-5和Pd/Pr-CeO2-10催化剂反应后的TEM和HRTEM如支持信息图S5所示, TEM结果表明催化剂的纳米棒结构在反应过程中保持较好. 且两种催化剂中均没有观察到明显的纳米颗粒.

图3 (a) Pd/Pr-CeO2-2.5催化剂的TEM图像和(b) HRTEM图像; (c) Pd/Pr-CeO2-5催化剂的TEM图像和(d) HRTEM图像; (e) Pd/Pr-CeO2-7.5催化剂的TEM图像和(f) HRTEM图像; (g) Pd/Pr-CeO2-10催化剂的TEM图像和(h) HRTEM图像Figure 3 (a) TEM image and (b) HRTEM image of Pd/Pr-CeO2-2.5 catalyst; (c) TEM image and (d) HRTEM image of Pd/Pr-CeO2-5 catalyst; (e) TEM image and (f) HRTEM image of Pd/Pr-CeO2-7.5 catalyst; (g) TEM image and (h) HRTEM image of Pd/Pr-CeO2-10 catalyst |
电感耦合等离子发射光谱(ICP-OES)测量的Pd/Pr-CeO2-2.5, Pd/Pr-CeO2-5, Pd/Pr-CeO2-7.5和Pd/Pr-CeO2-10的实际Pd含量分别为0.42%, 0.35%, 0.44%, 0.41%, 并且CO脉冲吸附测定结果表明, 所有催化剂中Pd都具有很高的分散度(表1). 为了观察Pr-CeO2载体上Pd物种和其他元素的分散情况, 我们对催化剂进行了高角环形暗场扫描透射电子显微镜(HAADF-STEM)和能量色散X射线光谱(EDX)元素图谱的表征. 如支持信息图S6所示, 可以看出Pd、Pr、Ce和O均匀分布在所制备纳米棒上, 进一步表明Pr成功掺杂到CeO2纳米棒的结构中形成固溶体, Pd也被成功负载在纳米棒上, 且Pd的分散较好.
表1 Pd/Pr-CeO2催化剂中各金属质量分数及Pd分散度Table 1 Each metal mass fraction and Pd dispersion of Pd/Pr-CeO2 catalyst |
| 催化剂 | w(Pd负载量)/% | w(Ce)/% | w(Pr)/% | Pd分散度/% |
|---|---|---|---|---|
| Pd/Pr-CeO2-2.5 | 0.42 | 56.26 | 1.26 | 98.46 |
| Pd/Pr-CeO2-5 | 0.35 | 50.75 | 2.48 | 97.52 |
| Pd/Pr-CeO2-7.5 | 0.44 | 53.39 | 3.75 | 97.14 |
| Pd/Pr-CeO2-10 | 0.41 | 51.42 | 5.10 | 98.67 |
各金属的质量分数通过ICP-OES测得; Pd分散度由CO脉冲吸附测定. |
2.3 催化剂氧空位表征
所有催化剂的Raman光谱如图4a所示, 在455 cm−1处出现的强峰可以归因于二氧化铈纳米棒立方萤石型结构的F2g振动模式[31-32]. 由于Pr3+的离子半径(0.113 nm)比Ce4+的离子半径(0.087 nm)大, Pr的掺杂引起了CeO2晶格的畸变, 离子半径的差异导致Pr-CeO2的F2g能带的位置和强度发生了一定的变化. 570 cm−1附近较弱的峰归因于CeO2表面的氧空位(Ov). I570/I455可以定量描述催化剂中氧空位的浓度. 通过计算得出, 四种催化剂中的氧空位浓度遵循以下顺序: Pd/Pr-CeO2-5 (0.60)>Pd/Pr-CeO2-7.5 (0.41)>Pd/Pr-CeO2-10 (0.32)>Pd/Pr-CeO2-2.5 (0.26). 适量的Pr3+离子掺杂到CeO2纳米棒中会产生更多氧空位, 以补偿 Pr3+离子的低价掺杂引起的电荷不平衡, 从而导致Ce3+浓度的增加.
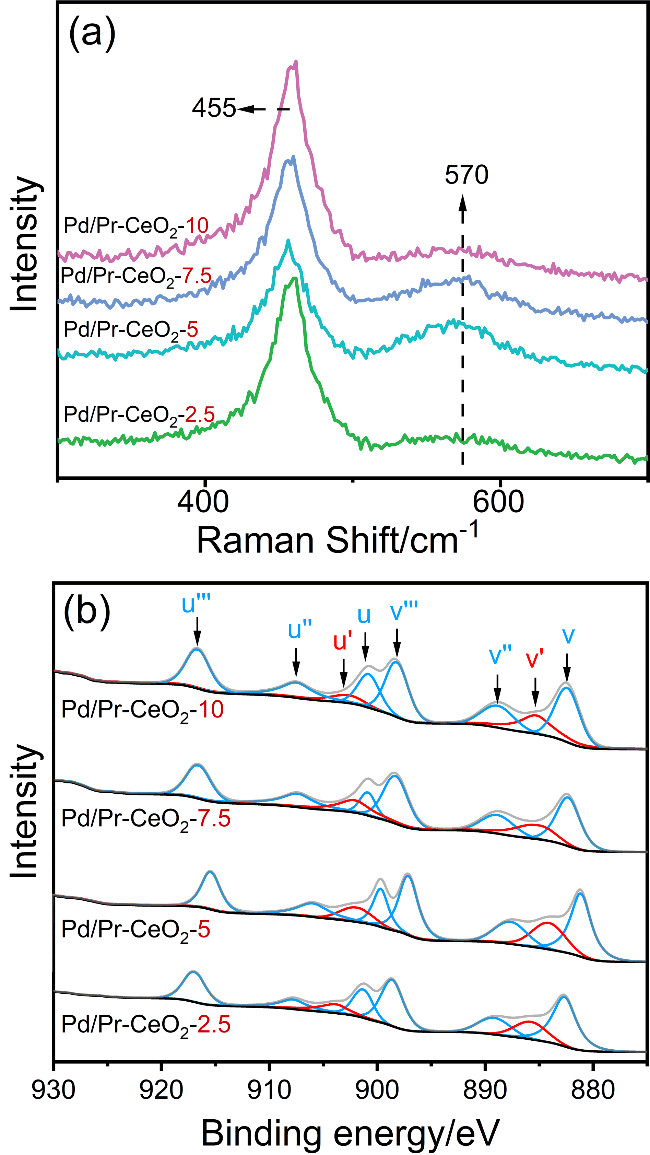
进一步通过XPS表征了其氧空位浓度, 各种Pr-CeO2载体和Pd/Pr-CeO2催化剂的Ce 3d XPS光谱分别如支持信息图S7和图4b所示. Ce 3d XPS光谱被反卷曲成8个峰, 结合能为v (882.5 eV)、v' (885.1 eV)、v" (888.6 eV)、v''' (898.4 eV)的四组峰归属于Ce 3d5/2自旋轨道. 而在结合能为u (900.9 eV)、u' (903.6 eV)、u" (907.3 eV)、u''' (916.7 eV)的四组峰归属于Ce 3d3/2自旋轨道[29,33-34]. 其中v'和u'归属于Ce3+物种, 其余6个峰归属于Ce4+物种. 载体和催化剂表面的Ce3+含量通过XPS数据中的峰面积来获得: Ce3+/(Ce3++Ce4+). Pd/Pr-CeO2催化剂的Ce 3d光谱计算结果表明, 四种催化剂中Ce3+含量遵循以下顺序: Pd/Pr-CeO2-5 (18.8%)>Pd/Pr-CeO2-7.5 (16.5%)>Pd/Pr-CeO2-10 (12.9%)>Pd/Pr-CeO2-2.5 (12.7%). 说明在载体中掺入适量的Pr可以增加其表面氧空位, 但掺入过多会导致氧空位减少.该结果与拉曼光谱的结果具有一致性. 氧空位含量的增加是因为适量Pr掺入可以有效降低CeO2 (111)晶面的氧空位形成能, 氧空位更容易产生[35]. 支持信息图S8是反应后催化剂的Ce 3d XPS谱图, 反应后催化剂中Ce3+含量遵循以下顺序: Pd/Pr-CeO2-5 (21.0%)>Pd/Pr-CeO2-7.5 (18.2%)>Pd/Pr-CeO2-10 (16.4%)>Pd/Pr-CeO2-2.5 (13.4%). 值得注意的是, 与新鲜催化剂相比, 反应后各催化剂中的Ce3+含量均有所增加, 但总体顺序保持一致. Ce3+含量的增加可能是催化剂表面的Ce4+在反应中被CO气体部分还原导致的.
2.4 Pd与载体相互作用和价态表征
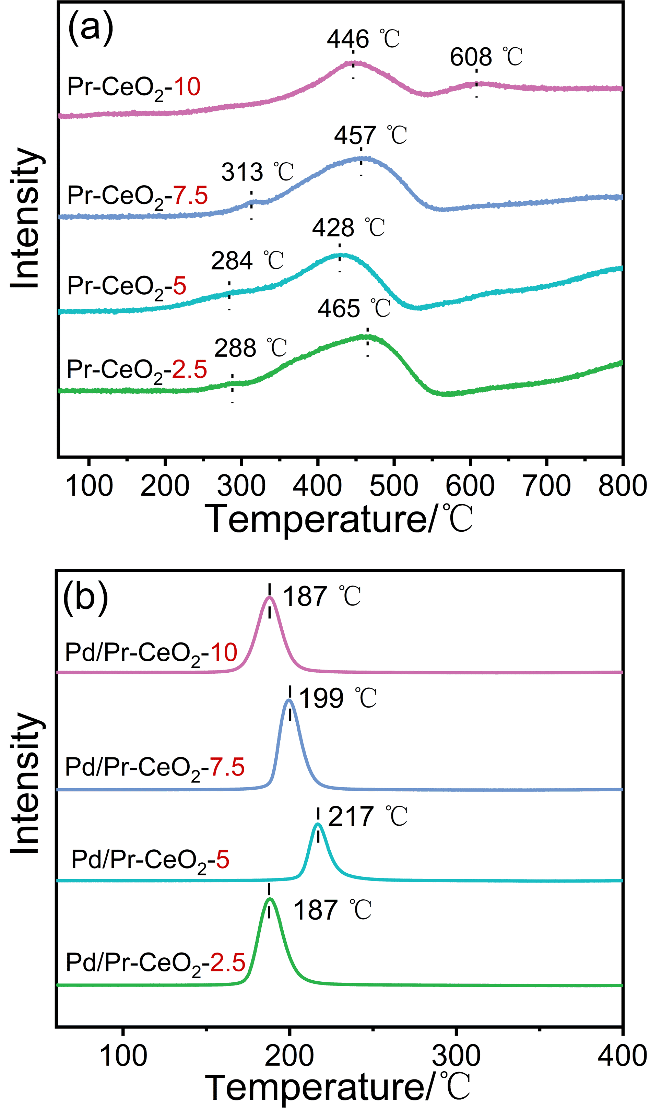
进一步利用H2-TPR来研究Pd活性物质和载体之间的相互作用. 如图5所示, 制备的载体在250~500 ℃存在一个耗氢区, 归因于纳米棒表面氧物种的还原. 而高于550 ℃的其他峰可能归因于CeO2还原为Ce2O3[26]. 在催化剂程序升温还原的过程中, 还原峰集中在150~250 ℃, 主要归因于PdOx的还原, 这是Pd在载体表面高度分散的特征. 随着Pr掺杂量的增加, PdOx的还原温度先升高后降低, 表明Pd与Pr-CeO2载体之间的金属-载体相互作用先增强后减弱. 在Pd/Pr-CeO2-5中, 其还原温度最高为217 ℃, 说明载体Pr-CeO2-5与活性组分Pd的相互作用最强, Pd更难以被还原成金属颗粒.
众所周知, 在CO酯化制DMC反应中, 催化剂中Pd活性组分的价态对产物选择性有巨大的影响, Pd(0)更有利于产生DMO, 而Pd(Ⅱ)更有利于产生DMC[17]. 因此我们通过XPS表征对反应前后催化剂中Pd的价态进行了分析, 图6a为反应前催化剂中Pd的3d轨道XPS图谱, 在338 eV和343 eV左右处可观察到两个特征峰, 分别对应Pr-CeO2上PdOx物种或PdxCe1-xO2-σ固溶体中的Pd2+. 图6b为反应后催化剂中Pd的3d轨道XPS图谱. 在反应后的催化剂Pd的3d轨道XPS图谱中我们观察到了Pd(0)的出现, 说明在反应中有部分Pd(Ⅱ)不稳定, 在反应中发生还原, 催化剂反应后表面Pd(Ⅱ)所占比例遵循以下顺序: Pd/Pr-CeO2-5 (74%)>Pd/Pr- CeO2-7.5 (62%)>Pd/Pr-CeO2-10 (36%)>Pd/Pr-CeO2-2.5 (33%). 因此Pd/Pr-CeO2-5具有最高的DMC选择性, 同时也说明Pd在Pr-CeO2-5载体上最难被还原, 该结果与H2-TPR的结果具有一致性.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
综上所述, 我们通过调节Pr元素的掺杂量制备了一系列Pr-CeO2载体, 并通过浸渍法负载Pd, 后续用于CO直接酯化制DMC的反应中, 发现其催化选择性存在显著差异. 首先通过XRD、TEM和ICP的表征, 证明Pr元素成功地引入载体中. 之后通过XPS和Raman光谱的表征, 发现合成的载体和催化剂中的氧空位浓度不同, DMC选择性的变化趋势与氧空位浓度变化趋势保持一致. 结合对Pd 3d XPS的表征和H2-TPR结果表明, 金属与载体之间的相互作用不同. 氧空位的引入增强了金属与载体间的相互作用, 有利于保持Pd(Ⅱ)高度分散而不易被反应气中的CO还原, 更有利于选择性生成DMC. 该工作揭示了CeO2载体的氧空位浓度与DMC选择性之间的构-效关系, 对于研发非分子筛体系的高性能DMC催化剂具有重要指导意义.
4 实验部分
4.1 催化剂制备
载体材料采用水热法制备: 称取19.2 g NaOH溶解在70 mL去离子水中得到溶液A, 称取不同物质的量比的Ce(NO3)3•6H2O和Pr(NO3)3•6H2O溶解于10 mL去离子水中得到溶液B. 将得到的溶液B缓慢地加入到溶液A中并在室温下以400 r/min搅拌30 min. 将混合物转移到反应釜中, 在100 ℃的恒温烘箱中反应约24 h, 取出待冷却至室温, 用离心机分离样品, 用乙醇和去离子水分别洗涤下层沉淀物数次. 之后将洗涤好的样品置于60 ℃恒温箱中干燥过夜, 最后将粉末在400 ℃的空气中煅烧2 h, 即可制得不同比例Pr掺杂的CeO2载体, 分别记作Pr-CeO2-X (X: 2.5, 5, 7.5, 10).
Pd/Pr-CeO2-X催化剂采用浸渍法制备: 首先量取1.5 mL Pd(NO3)2溶液(2.17 mg/mL)分散于15 mL去离子水中形成淡黄色溶液, 再将0.3 g上述制备的Pr-CeO2-X载体加入到溶液中, 缓慢加热至溶液全部蒸发, 整个过程需要在磁力搅拌下进行, 最后得到固化物. 收集固化物至坩埚中, 先在110 ℃下干燥2 h, 后在300 ℃的马弗炉中空气气氛下焙烧4 h, 即得到0.5% (w) Pd理论负载量的Pd/Pr-CeO2-X催化剂(X: 2.5, 5, 7.5, 10).
4.2 催化性能测试
在固定床石英管反应器中进行CO酯化制DMC催化剂的活性评价. 在石英管式反应器中间放置200 mg催化剂, 将原料气(体积分数为19% CO, 45% CH3ONO, 3% Ar作为内标, 33% N2作为平衡气)以2500 L•kgcat-1•h-1的空速通过催化剂, 设定反应温度为120 ℃, 并维持系统压力约为0.1 MPa, 采用配备热导检测器和氢火焰离子化检测器的岛津GC-2014气相色谱在线监测反应物和产物的组成.
使用以下公式计算CO转化率和对DMC选择性:
(1) 基于内标法计算CO转化率
CO转化率(%)=([CO]in/[Ar]in-[CO]out/[Ar]out)/ ([CO]in/[Ar]in)×100%
其中[CO]in和[CO]out分别指反应前后CO的峰面积, [Ar]in和[Ar]out分别指反应前和反应后混合气体中氩气的峰面积. [CO]in和[Ar]in通过按照比例单独通入反应气体的空管数据采集所得.
(2) 基于CO计算产物的选择性
DMC选择性(%)=(SDMC×R-FDMC/MDMC)/(SDMC×R-FDMC/MDMC+2×SDMO×R-FDMO/MDMO)×100%
DMO选择性(%)=(2×SDMO×R-FDMO/MDMO)/(SDMC×R-FDMC/MDMC+2×SDMO×R-FDMO/MDMO)×100%
其中SDMC和SDMO分别代表碳酸二甲酯(DMC)和草酸二甲酯(DMO)的峰面积, R-FDMC和R-FDMO分别对应其相对校正因子, MDMC和MDMO分别代表相对分子量.
(Cheng, B.)