

1 引言
2 结果与讨论
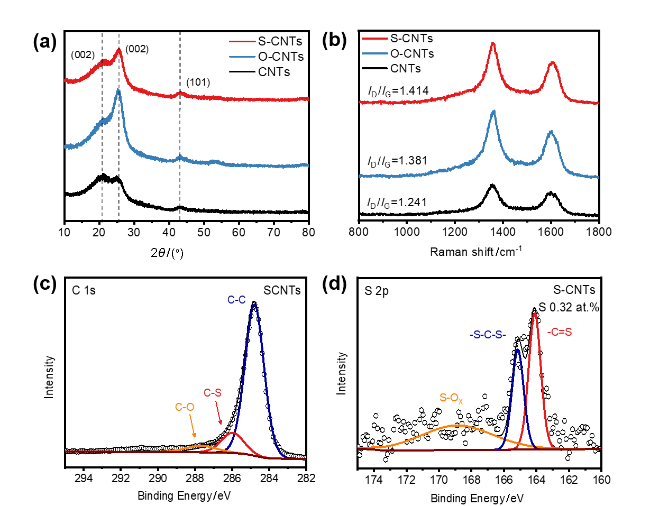
2.1 物理表征分析

图1 S-CNTs的合成示意图(a); S-CNTs的TEM (b, c和d)和HRTEM (e, f)图像, (f)为(e)方框区域的放大图; S-CNTs的高角度环形暗场(HAADF)和相应的元素能谱分析(EDS) mapping图像(g)Figure 1 Schematic representation of the synthesis of S-CNTs (a); TEM (b, c and d), and HRTEM (e, f) images of S-CNTs, (f) is a zoomed-in view of the boxed area in (e); High-angle annular dark field (HAADF) and corresponding elemental energy-dispersive X-ray spectroscopy (EDS) mapping images of S-CNTs (g) |
2.2 电化学性能分析
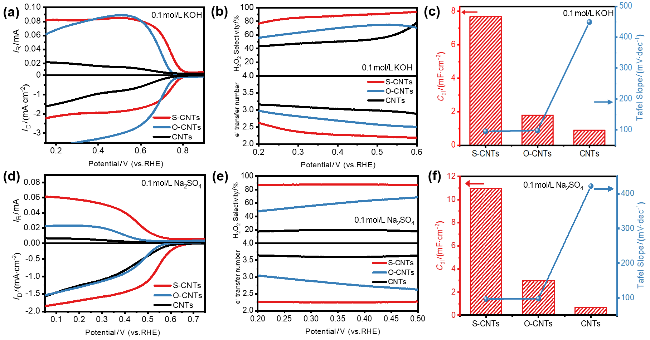
图3 S-CNTs催化剂在RRDE测试的LSV曲线(0.1 mol•L-1 KOH溶液)(a), 电子转移数和选择性(b), CNTs, O-CNTs和S-CNTs催化剂的Tafel Slope和Cdl对比(c); S-CNTs催化剂在RRDE测试的LSV曲线(0.1 mol•L-1 Na2SO4溶液)(d), 电子转移数和选择性(e), CNTs, O-CNTs和S-CNTs催化剂的Tafel Slope和Cdl对比(f)Figure 3 LSV curves of S-CNTs catalysts tested at RRDE (0.1 mol•L-1 KOH) (a), electron transfer number and selectivity (b), Tafel Slope and Cdl comparisons of CNTs, O-CNTs and S-CNTs catalysts (c); LSV curves of S-CNTs catalysts tested at RRDE (0.1 mol•L-1 Na2SO4) (d), electron transfer number and selectivity (e), Tafel Slope and Cdl comparisons of CNTs, O-CNTs and S-CNTs catalysts (f) |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图4 S-CNTs催化剂在碱性(1 mol•L-1 KOH)三电极流动池测试的LSV曲线(a), 法拉第效率和产率(b); S-CNTs催化剂在中性(1 mol•L-1 Na2SO4)三电极流动池测试的LSV曲线(c), 法拉第效率和产率(d)Figure 4 LSV curves (in 1 mol•L-1 KOH solution) (a), Faradaic efficiency and yield (b) for S-CNTs catalysts tested in alkaline three-electrode flow cell; LSV curves (in 1 mol•L-1 Na2SO4) (c), Faradaic efficiency and yield (d) for S-CNTs catalysts tested in neutral three-electrode flow cell |