

1 引言
2 结果与讨论
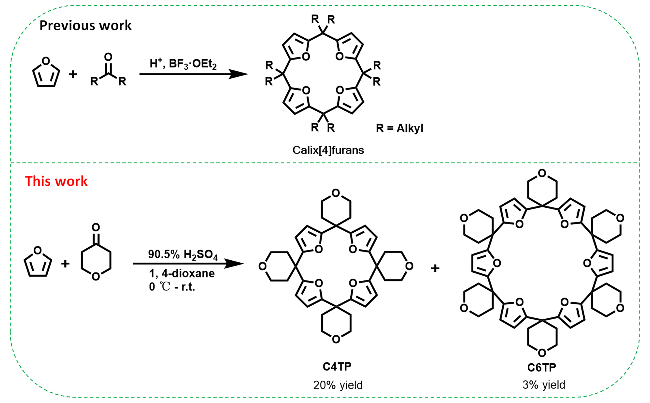
2.1 杯呋喃大环化合物合成
2.2 杯呋喃大环C4TP和C6TP的核磁共振氢谱(1H NMR)和质谱(MS)
2.3 杯呋喃大环C4TP和C6TP晶体结构和堆积

2.4 杯呋喃大环C4TP和C6TP的物理化学性质
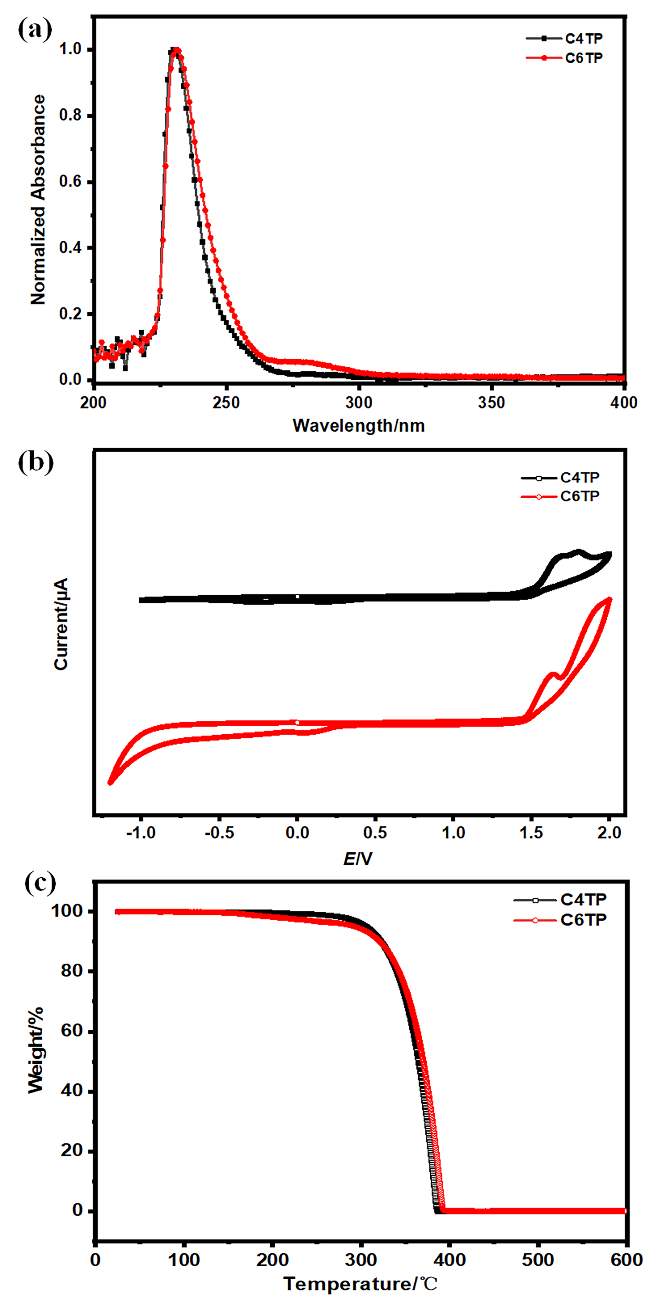
图3 C4TP和C6TP的(a)紫外-可见光谱图, (b)循环伏安法电化学图和(c)热重分析图Figure 3 (a) UV-Vis spectra, (b) cyclic voltammograms (CV) and (c) thermogravimetric analysis (TGA) of C4TP and C6TP |
表1 C4TP, C6TP和C4TP-MSA的光物理和电化学数据Table 1 Optical and electrochemical data for C4TP, C6TP and C4TP-MSA |
| UV-Visa | Egopt/ eV | λemib/ nm | Stokes/ nm | Td c/ ℃ | CVd | |
|---|---|---|---|---|---|---|
| λmax/nm | Eox/V | |||||
| C4TP | 231 | 5.37 | 352 | 122 | 306 | 1.50 1.48 — |
| C6TP | 231 | 5.37 | — | — | 293 | |
| C4TP-MSA | 615 | 2.02 | 516 | — | — |
a The photophysical properties of the compounds were measured in CH2Cl2 (C4TP 10−4 mol•L−1), n(C4TP)∶n(MSA)=1∶1000 (molar ratio); Egopt is the optical band gap and estimated from the onset of the absorption peak, (Egopt=1240/λonset); b Measured in CH2Cl2 (concentration of C4TP is 10−6 mol•L−1); c Decomposition temperature determined by TGA corresponding to 5% weight loss at 10 ℃•min−1 under nitrogen flow. d CVs were measured in ultradry dichloromethane with TBAPF6 (0.1 mol•L−1) as the supporting electrolyte and a scan rate of 100 mV•s−1; Eox is the onset of the oxidation potential. |
2.5 杯呋喃大环C4TP与酸相互作用研究
2.5.1 杯呋喃大环C4TP与系列酸作用体系的紫外-可见(UV-Vis)吸收光谱
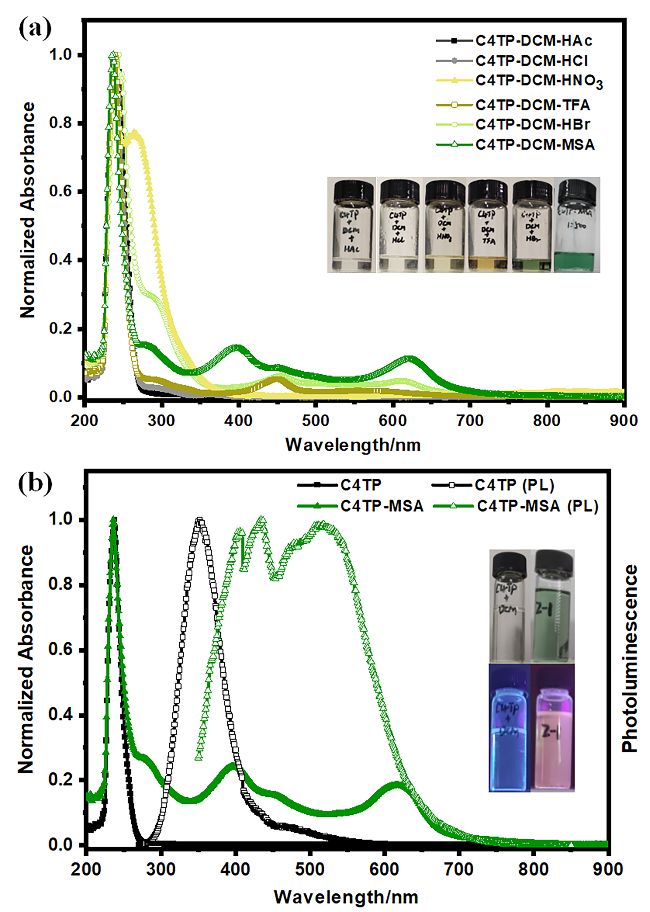
图4 (a)大环C4TP与酸分子(HAc, HCl, HNO3, TFA, HBr, MSA)作用体系的紫外-可见吸收光谱, 内插图为大环C4TP与酸分子作用体系的溶液在日光下的颜色. (b)大环C4TP与C4TP-MSA作用体系的归一化紫外与荧光光谱, 内插图: 左上和右上分别为C4TP和C4TP-MSA溶液在日光下的颜色, 左下和右下分别为C4TP和C4TP-MSA溶液在365 nm紫外光照射下的颜色Figure 4 (a) UV-Visible absorption spectra of macrocyclic C4TP interacting with acid molecules (HAc, HCl, HNO3, TFA, HBr, MSA). Insets: The solvent colors of C4TP-acid under daylight; (b) Normalized UV and fluorescence tests of the C4TP-MSA interaction system. Insets: The top left and top right show the colors of C4TP and C4TP-MSA solutions under daylight, respectively; the bottom left and bottom right show the colors of C4TP and C4TP-MSA solutions under 365 nm UV light, respectively |
2.5.2 杯呋喃大环C4TP与MSA作用体系的UV-Vis光谱

图5 (a)大环C4TP与MSA(物质的量比为1∶1000, 不同时间)作用体系的紫外-可见吸收光谱, 大环C4TP浓度为1.0×10−4 mol•L−1, 溶剂为二氯甲烷, (b) 390 nm和620 nm处峰值强度随时间变化图, (c)大环C4TP与MSA(物质的量比为1∶1000, 不同时间)作用体系溶液的颜色Figure 5 (a) The UV-Vis absorption spectra of the system where the macrocycle C4TP interacts with MSA (molar ratio as 1∶1000, at different times), with a concentration of C4TP at 1.0×10−4 mol•L−1 and the solvent being dichloromethane; (b) The plot of peak intensities at 390 nm and 620 nm along with time; (c) The color of the solution in the system where the macrocycle C4TP interacts with MSA (molar ratio as 1∶1000, at different times) |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图6 (a)大环C4TP与MSA(不同物质的量比, 以大环C4TP为单位1)作用体系的紫外-可见吸收光谱, 大环C4TP浓度为1.0×10−4 mol•L−1, 溶剂为二氯甲烷, (b) 390 nm和620 nm处峰值强度随增加酸物质的量的变化图, (c)大环C4TP与MSA (不同物质的量)作用体系的颜色Figure 6 (a) The UV-Vis absorption spectra of the system where the macrocycle C4TP interacts with MSA (at different equivalents), with a concentration of C4TP at 1.0×10−4 mol•L−1 with solvent of dichloromethane; (b) The plot of peak intensities at 390 nm and 620 nm along with increasing acid equivalents; (c) The color of the solution in the system where the macrocycle C4TP interacts with MSA (at different equivalents) |