

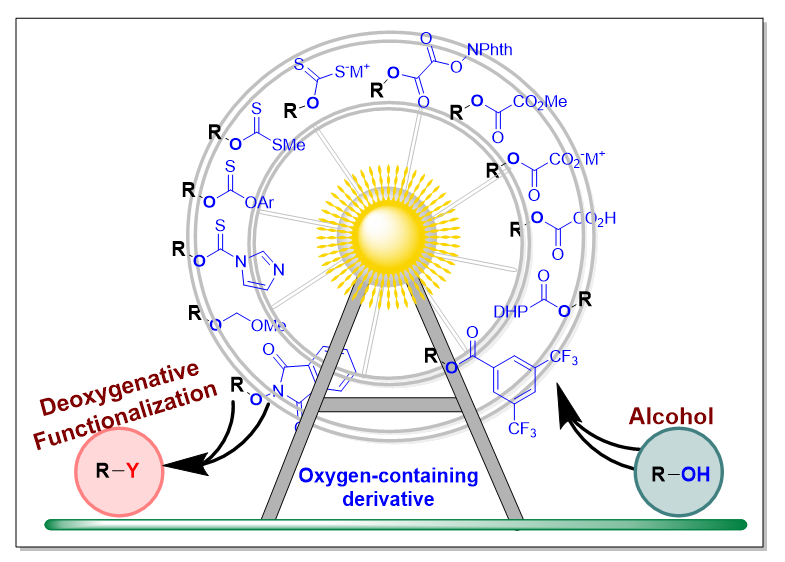
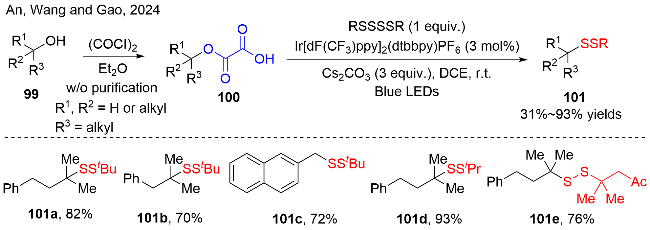
1 引言
醇类化合物广泛存在于自然界中, 在实验室里醇羟基也是有机官能团中最容易获取的基团之一. 醇的脱氧化反应是现代有机化学中非常重要的板块之一, 涉及的研究领域相当广泛. 例如, 在能源领域, 将有机原料转化为生物质燃料, 就像在天然产物合成中去除羟基一样. 大多数非选择性的方法是对自然界中的碳水化合物进行转化, 用以生产生物燃料, 但有机化学家们会以醇类化合物为原料, 通过化学选择性的脱氧反应来合成特定分子. 这种类型的脱氧化反应应该具有广泛的底物范围, 并可以兼容许多类型的官能团. 我们在此讨论了醇类化合物的脱氧化/功能化反应在有机合成中的应用.
在醇类化合物中, 由于醇中的C—OH键的解离能较高(约544到669 kJ/mol)[1], 所以有机合成中的羟基负离子是一个比较差的离去基团, 非活化的C—OH键的断裂是热力学上不支持的, 而且直接单电子还原醇羟基需要更高的还原电位. 因此, 直接切断醇中的C—OH键, 实现脱氧/官能团化是一项挑战性的任务. 一个可行的方法是将羟基转化为一个良好的离去基团或特殊结构的自由基前体进行反应. 我们总结了醇类化合物进行脱氧化反应的四种途径, 如图式1所示. 在传统的合成方法中, 醇的脱氧转化主要采用两步法实现: 一是在酸性条件下醇先发生消除反应生成烯烃[2], 再经历碳碳双键的功能化, 可以转化为其他有价值的化合物(图式1b); 二是将醇羟基转化为适当的离去基团(不含醇羟基中的氧原子, 例如: 卤素), 接着经历脱氯、功能化反应, 实现醇的脱氧转化(图式1c)[3]. 这也是有机合成中应用最为广泛的脱氧转化策略.

半个世纪前, Barton-McCombie脱氧反应被发现[4], 这是研究较为深远的一种醇的脱氧转化方法. 该方法是将醇直接转化为硫代羰基衍生物, 经历C—O键均裂产生烷基自由基中间体, 后续从Bu3SnH中攫取氢原子, 实现醇的脱氧/还原为烃. 随着自由基化学的快速发展, 为了避免使用过量的危险试剂和遭受苛刻的反应条件, 化学家们也对Barton-McCombie脱氧反应进行了持续改进[5]. 例如, 采用有机金属单电子试剂、电化学方法、光诱导的单电子转移过程, 或者可见光氧化还原催化的方法实现[6]. 在过去的十几年里, 可见光氧化还原催化成为一种强劲的有机合成新策略, 可以实现许多重要的化学转化[7]. 此时, 醇类化合物的直接脱氧转化在该领域也得到了快速的发展. 例如, 在有机磷化合物的存在下, 通过自由基的途径, 可以实现醇类化合物的直接脱氧转化, 已经有相关综述对其进行详细介绍(图式1a)[8]. 另外, 醇的含氧衍生物, 例如, 羧酸酯、草酸酯、黄原酸酯和醚类等化合物及其衍生物, 在可见光氧化还原催化的条件下, 通过C—O键的均裂得到烷基碳自由基中间体, 后续经历自由基加成、偶联或攫取原子等过程, 可以实现脱氧/功能化(图式1d).
在此, 我们主要针对过去十年里, 不同类型醇的含氧衍生物(可以分离出来), 在可见光化学反应条件下, 进行的脱氧/功能化反应的研究进展进行总结. 由于醇的含氧衍生物的结构不同, 导致催化C—O键断裂的机理过程不同, 它们离去的含氧基团也有所不同. 我们根据醇的含氧衍生物的结构不同, 包括羧酸酯、草酸酯、硫代碳酸酯和其衍生物、醚类化合物和其他的醇衍生物, 分为五部分进行阐述. 每一部分针对醇的衍生物的精细结构、反应的化学转化和催化方式进行展开, 详细讨论合成策略、化合物的重要性、反应机理以及反应的优缺点. 后续的合成应用也会呈现, 特别是天然产物、生物活性和药物活性分子的功能化反应. 在我们投稿之前, 也有其他团队[8f-8g]报道了和我们类似主题的研究进展, 这也进一步说明了化学家们对醇的脱氧化反应产生了浓厚的兴趣.
2 羧酸酯的脱氧/功能化反应
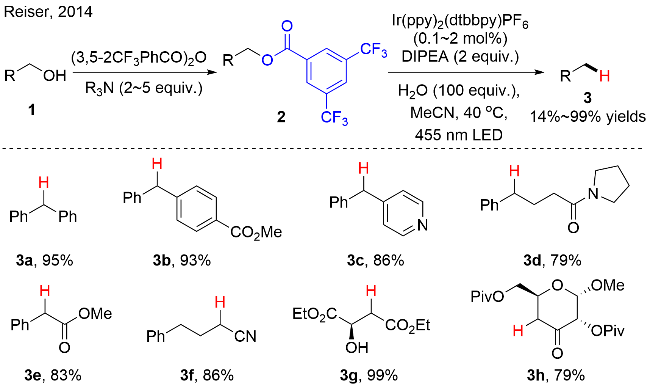
该芳基甲酸酯的脱氧/还原反应机理如图式3所示: 首先, IrIII在可见光的照射下, 转变成其激发态*IrIII. 三级胺作为还原猝灭剂与激发态*IrIII发生单电子转移过程, 产生还原性的IrII和三级胺自由基正离子中间体.
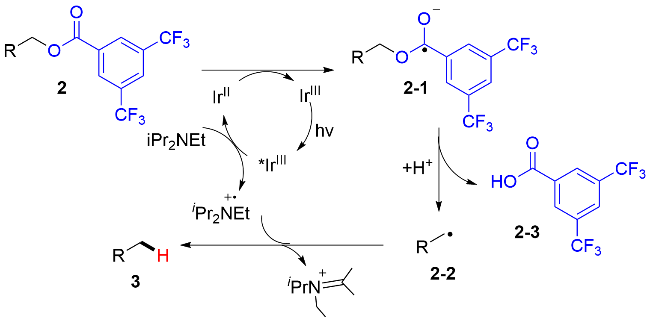
IrII将缺电子的芳基甲酸酯2还原, 回到基态IrIII, 同时产生中间体2-1, 该中间体通过C—O键均裂, 生成烷基碳自由基2-2和芳基甲酸2-3. 最后, 烷基碳自由基2-2从三级胺自由基正离子中夺取一个氢原子, 得到还原氢化的产物. 此外, 作者通过计算化学确认了中间体2-1结构的稳定性.
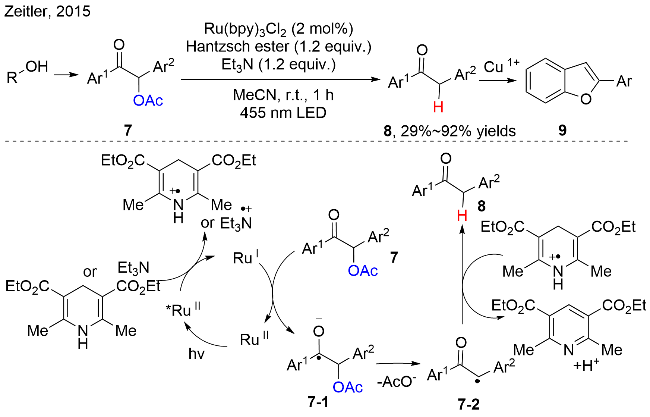
2015年, Zeitler等[12]报道了乙酰基取代的安息香衍生物7的脱氧/还原反应. 该反应是以金属钌为光催化剂, 在三乙胺和汉斯酯作为添加剂时, 在温和的条件下, 高效地制备了一系列芳基酮类化合物8(图式5). 控制实验表明: 当没有添加三乙胺时, 反应的产率会大大降低; 当无汉斯酯参与时, 基本无目标产物的生成; 当没有三乙胺和汉斯酯存在时, 即使延长时间该反应也没有目标产物的产生. 这些说明汉斯酯在反应中起到至关重要的作用, 既作为氢原子供体, 也作为还原猝灭剂参与反应; 当同时有三乙胺作为还原猝灭剂时, 可以快速提高反应的产率. 该反应的机理是化合物7经历单电子还原, C—O键发生单电子还原断裂, 脱去乙酰氧基负离子中间体, 得到关键的烷基碳自由基7-2. 此外, 化合物8通过铜催化的环化反应, 直接可以制备2-芳基取代的苯并呋喃化合物9.
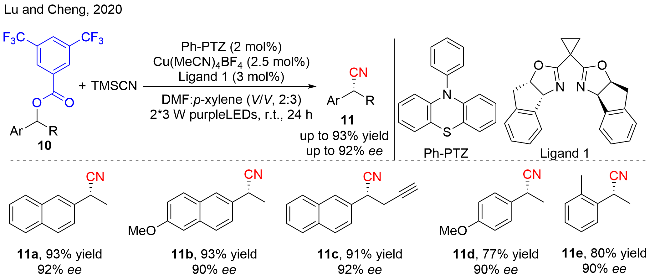
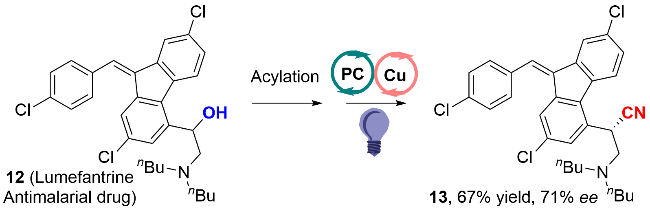
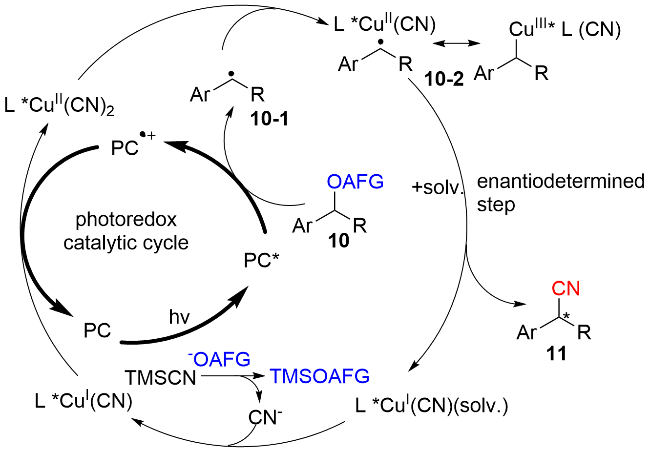
作者通过自由基钟反应证实了苄基自由基的存在, 量子收率(Φ=0.36)和灯开/关实验揭示了该反应以连续的光反应为主, 不存在自由基链反应. 作者提出了可靠的反应机理过程如图式8所示: 首先, 在紫光的照射下, PC激发到其激发态PC*, 随后被化合物10氧化, 产生氧化性的PC•+, 同时生成苄基自由基中间体10-1. 之后, 一价铜氰化物被PC•+氧化, 得到二价铜氰化物和基态的PC. 苄基自由基中间体10-1被二价铜捕获, 得到三价的铜复合物10-2, 随后发生还原消除, 获得最终的苄基腈化合物11. 这一步也是立体选择性的决定步骤. 在该策略中, 光催化促进了苄醇衍生物中C—O键的断裂, 而铜催化致使C—C键的生成.
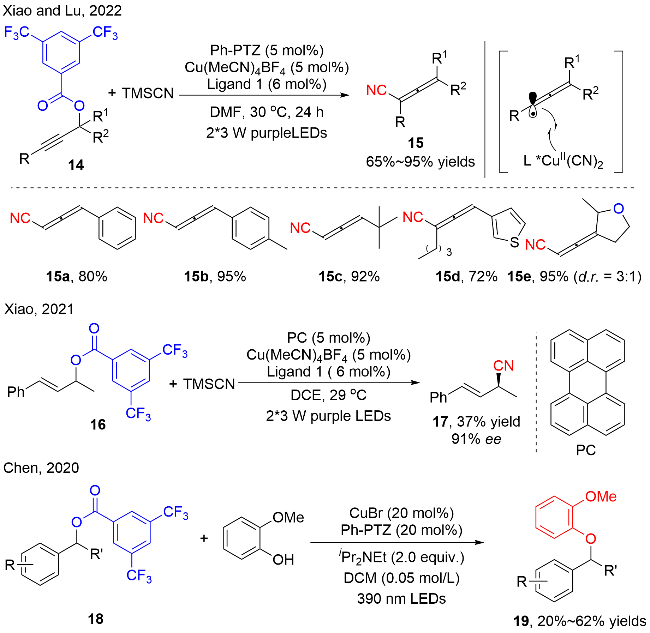
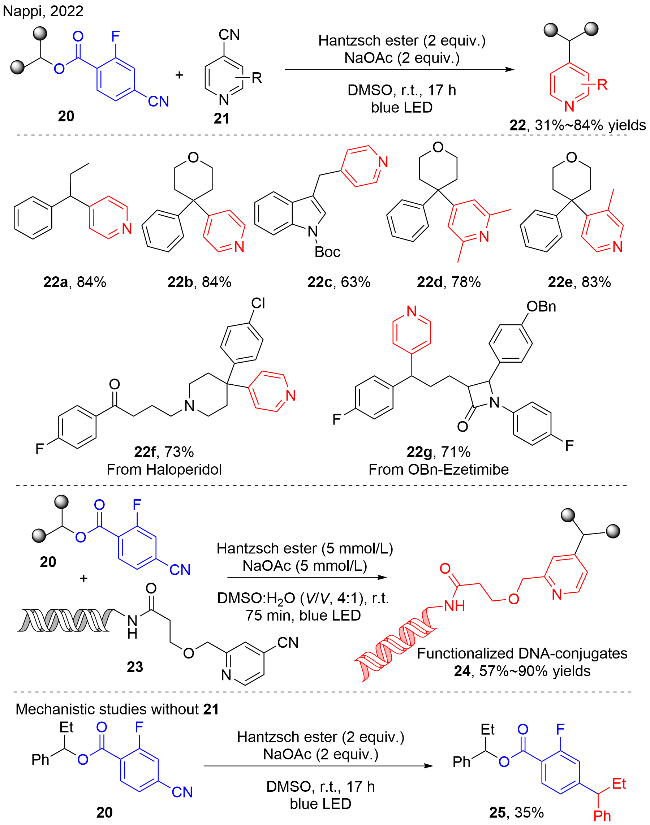
对于该反应的机理, 作者通过在标准反应中加入2,2,6,6-四甲基哌啶-1-氧自由基(TEMPO), 分离得到TEMPO与苄基碳自由基的加成产物, 证明有苄基碳自由基的产生; 紫外可见光谱实验暗示了汉斯酯(HE)与醋酸钠之间存在相互作用, 向汉斯酯(HE)与醋酸钠体系中加入化合物20, 光谱没有明显的变化; 但是Stern- Volmer猝灭实验显示HE/醋酸钠体系的荧光可以被化合物20猝灭, 说明它们之间存在电荷转移过程. 此外, HE (Ered=-2.28 V vs. SCE)也能够独自与4-氰基吡啶21 (Ered=-1.87 V vs. SCE)发生单电子还原, 产生21的自由基阴离子物种. 这些机理实验暗示了该反应是通过烷基自由基与4-氰基吡啶自由基阴离子中间体进行交叉偶联的方式提供了目标产物22. 当模型反应中没有4-氰基吡啶参与时, 分离出化合物25, 这也进一步证实了反应的机理途径.
4-位取代1,4-二氢吡啶化合物(4-位取代的汉斯酯化合物)是一类重要的自由基前体, 在直接光激发或者光催化剂的存在下, 产生烷基碳自由基、芳酰基自由基或者氨基甲酰基自由基中间体, 进而进行各种功能化反应. C-芳基糖苷类化合物作为药物候选物得到了广泛研究. 2021年, Diao等[16]以取代的分子内半缩醛26为底物, 与羧酸27进行酯缩合反应, 得到4位-烷氧基酰基取代的1,4-二氢吡啶(DHP)化合物28, 并将其应用于光催化与镍协同催化的偶联反应中, 用来制备C-(杂)芳基取代的核苷和脱氧糖(图式11). 该反应的底物范围广泛, 官能团的兼容性良好, 不足之处在于反应需要在较高的温度下进行.
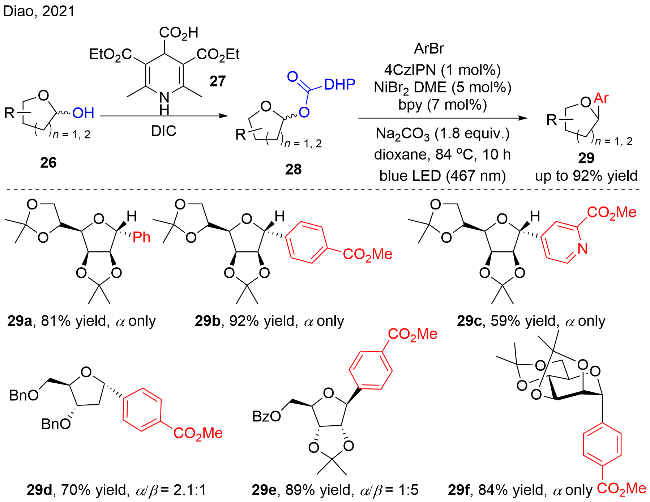
作者针对光催化与镍协同催化C-(杂)芳基取代糖苷的合成反应提出了可能的机理, 如图式12所示. DHP衍生的羧酸酯化合物28被激发态的PC*氧化, 发生C—C键和C—O键的断裂, 释放出CO2和吡啶副产物, 以及碳自由基中间体28-1和PC•−. 在Ni催化循环中, 零价镍复合物与芳基溴化物发生氧化加成, 得到二价镍复合物. 随后, 碳自由基28-1被二价镍复合物捕获, 生成三价镍复合物, 之后经历还原消除, 释放出芳基化的产物29和一价镍复合物. 最后, 一价镍复合物与低价态的PC•−发生单电子还原, 又回到零价镍, 同时光催化剂回到其基态PC, 进入下一个催化循环.
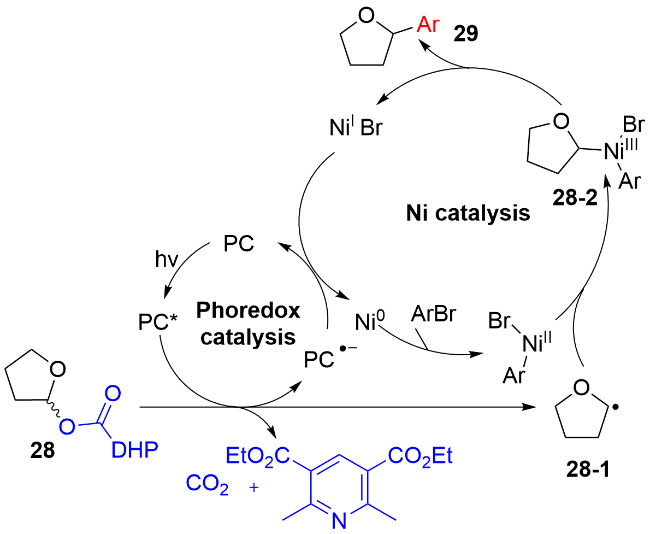
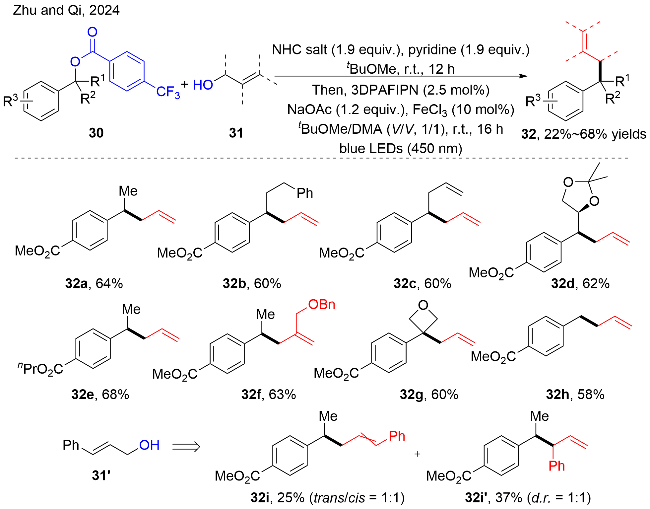
作者通过TEMPO和自由基加成环化反应证明苄基自由基的存在; 通过使用反式烯丙醇化合物31'时, 可以得到顺反比例相等的混合物32i和32i', 说明反应中存在烯丙基碳自由基. 根据这一系列实验作者提出了可行的反应途径, 如图式14所示. 首先, 氮杂卡宾盐与烯丙醇31进行反应, 生成氮杂卡宾(NHC)-烯丙醇的加成物31-1. 在可见光的照射下, 该加成物31-1作为还原剂猝灭激发态的PC*, 生成氮自由基阳离子物种31-2和低价态的PC•−. 接着, 该氮自由基阳离子物种31-2经历α位脱氢和C—O键断裂, 释放出氮杂卡宾的氧化物和烯丙基碳自由基. 同时, 芳基甲酸苄酯30在Fe3+的作用下, 被低价态的PC•−单电子还原, 脱去芳基甲酸铁盐, 生成苄基自由基中间体. 最后, 烯丙基自由基与苄基自由基发生自由基-自由基交叉偶联, 生成目标产物32. 作者通过测定该反应的量子产率(Φ=0.31), 排除了自由基链传递的过程.
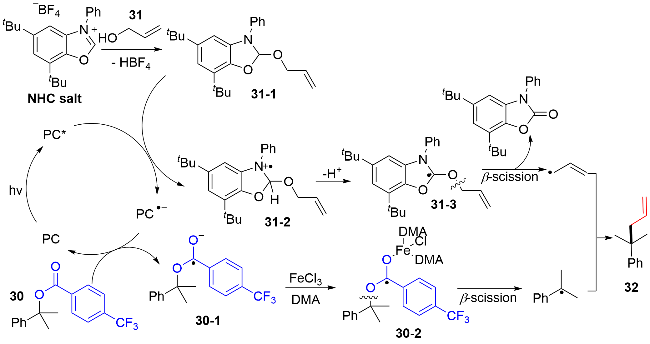
自由基-自由基交叉偶联是有机合成中一种高效的构建C(sp3)—C(sp3)键的途径, 但是对于通过C—O键和 C—H键的自由基-自由基交叉偶联反应报道的较少. 2024年, Xia和Chen等[18]发展了一种可见光氧化还原催化和氢键作用直接促进杂芳基苄醇衍生的芳基甲酸酯与甘氨酸衍生物的脱氧偶联反应的方法, 用于对含氨基酸片段的复杂结构的生物活性分子的制备(图式15). 该方法具有广泛的底物范围, 包括一级、二级和三级醇, 以及氨基酮、肽链和一些含手性辅助剂的甘氨酸衍生物都能较好的适应, 同时该反应的官能团的兼容性也很好. 作者通过控制实验证明: 磷酸的存在对该反应的收率起到至关重要的作用, 即在反应的过渡态中, 磷酸通过双氢键形式拉近氮杂芳甲基与二级胺化合物的空间距离, 提高了自由基与自由基发生交叉偶联过程的选择性.
3 草酸酯的脱氧/功能化反应
叔醇因其容易制备和商业廉价易得的优势而被视为一种理想的叔碳自由基前体. 然而, 由于其C—O键具有较高的解离能, 叔醇往往需要进行预先官能化, 即先转化为叔醇衍生物, 进而转化为有效的叔碳自由基前体[19]. 在可见光光氧化还原催化的条件下, 草酸烷基酯已经发展成为一类新型、稳定的烷基自由基前体化合物. 在2013年, Overman 等[20a]报道了一种由三级醇衍生的草酸酯36与缺电子烯烃37的偶联反应, 从而实现了由三级醇类衍生物向季碳化合物的快速构建(图式16). 反应底物草酸酯36可经由廉价易得的叔醇与草酰氯单酯(由草酰氯与N-OH邻苯二甲酰亚胺制备)进行反应获得. 该反应能够兼容一系列环状、链状的叔烷基草酸酯, 以及不同取代基的缺电子烯烃, 并能以高达92%的收率获得脱氧/官能团化产物38. 控制实验表明: 无二异丙基乙胺(DIPEA)的HBF4盐时, 目标产物的产率会大大降低. HBF4的作用: 一方面是使质子化的化合物36, 容易被光催化剂PC*单电子还原, 另一方面提供BF4−离子, 通过阴离子交换, 形成溶解性更好的Ru(bpy)3(BF4)2.

作者提出了一个可能的反应机理, 如图式16所示. 首先, 在可见光的照射下, RuII光催化剂由其基态转化为激发态, 随后, 与HE经过还原淬灭过程, 生成RuI光催化剂和氮自由基阳离子中间体HE-1. 紧接着, 还原态RuI光催化剂对草酸酯36进行单电子还原, 生成自由基阴离子中间体36-1. 该中间体36-1随后经连续两次的脱羧过程生成碳自由基中间体36-3. 缺电子烯烃37对碳自由基36-3进行捕获, 随后从HE-1夺氢, 生成目标产物38. 2015年, Overman团队[20b]采用类似策略实现了叔醇衍生物分别与烯丙基卤化物、乙烯基卤化物的还原偶联反应, 进一步拓展了该策略中缺电子烯烃的适用范围(图式16).
与在上述还原偶联策略不同的是, 单烷基取代的草酸盐作为醇的含氧衍生物, 也常作为烷基自由基前体被广泛使用. 2015年, MacMillan和Overman团队[21]发展了一种以仲、叔醇衍生的草酸铯盐作为烷基自由基前体, 与缺电子烯烃进行的偶联反应(图式17). 类似地, 该反应能够兼容一系列环状、链状和一些复杂结构的烷基草酸盐以及不同类型取代的缺电子烯烃, 并普遍能以较高的收率获得烯烃加成产物. 在该反应中, 叔醇衍生的烷基草酸铯44是一类结构比较稳定, 并且容易操作的合成中间体, 其经历单电子氧化和两次脱羧, 为叔醇向叔碳自由基的转化提供了一条温和、简便的途径. 值得注意的是, 当以叔醇47为底物, 经历中间体草酸盐48, 与烯烃49发生光化学反应, 最终能够以98%的收率获得二萜类天然产物反式克罗登烷50. 这也进一步证实了该合成策略的实用性.

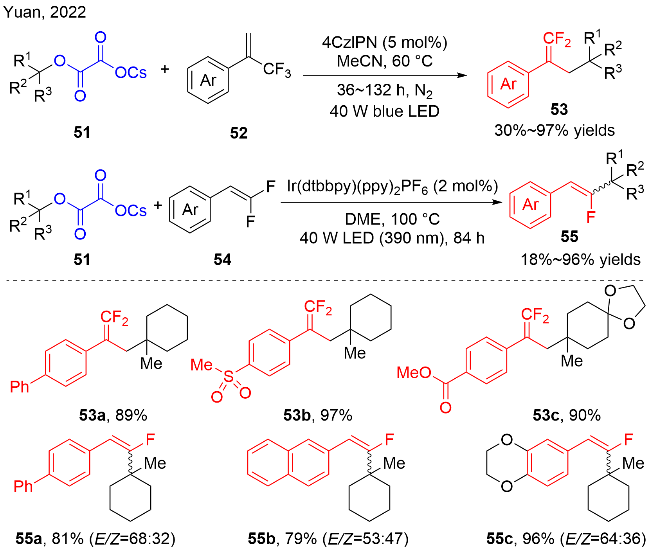
2022年, Yuan等[22]也采用烷基取代草酸盐51为烷基自由基前体, α-三氟甲基取代的烯烃52作为自由基受体进行加成反应, 实现了二氟烯丙基取代的季碳中心的构建(图式18). 在该反应中, 廉价易得的有机染料4CzIPN被证明是最优的光敏剂. 尽管需要较高的温度和较长的反应时间, 但是该反应能够很好地兼容不同类型的α-三氟甲基取代的芳基烯烃, 并能够以30%~97%的收率得到目标产物53. α-三氟甲基取代的烷基烯烃与2-三氟甲基-1,3-烯炔也能适用于该反应, 但收率有所下降(30%和59% yields). 当二氟代芳基烯烃54作为自由基受体时, 使用贵金属铱配合物作为光催化剂, 反应能以18%~96%的收率获得单氟代烯烃55. 然而, 更加苛刻的反应条件, 如: 更高的反应温度和更短波长的光源, 是必需的.
C(sp3)—C(sp2)键的构建也是一种增长碳链的有效手段. 2016年, MacMillan等[23]首次实现了可见光驱动光氧化还原催化与镍协同催化烷基取代草酸56与芳基溴代物的交叉偶联反应(图式19). 在该反应中, 醇与草酰氯反应生成的烷基取代的草酸56作为有效的烷基自由基前体, 芳基卤代物作为偶联对象, 经过双催化模式下, 实现了C(sp3)—C(sp2)键的构建. 一系列廉价易得的环状、链状伯醇与仲醇, 普遍能以中等至较好的收率得到偶联产物58. 同时, 该反应对芳基溴代物和杂芳基溴代物表现出很好的兼容性, 例如, 向苯环或吡啶环上不同位置引入不同类型的吸电子基或给电子基, 反应的收率并未受到明显的影响. 此外, 作者以简单、便宜易得的二级醇59为初试原料, 经历酰化、双催化醇的脱氧偶联反应、脱Boc和芳基的胺化反应, 得到化合物60, 该化合物是制备治疗肺结核的候选药物61的关键中间体.

光催化与镍协同催化烷基取代草酸的芳(氮杂)基化反应的机理历程, 如图式20所示. 首先, 在蓝光的照射下, IrIII被激发到其激发态*IrIII (E1/2red[*IrIII/IrII]=+1.1 V vs. SCE in MeCN), 随后与烷基取代草酸56 (Ep/2=+1.26 V vs. SCE in MeCN for the cesium salt)发生单电子转移, 得到IrII, 化合物56被单电子氧化、脱羧, 生成烷基自由基. 在Ni催化循环中, Ni0与ArBr发生氧化加成, 生成ArNiBr复合物. 之后, 烷基自由基被ArNiBr复合物捕获, 得到NiIII复合物. NiIII复合物还原消除, 释放出产物58和NiI复合物. 最后, NiI复合物被Ir2+ (E1/2red[IrIII/IrII]=-1.42 V vs. SCE in MeCN)还原, 恢复到Ni0复合物和基态的IrIII催化剂. 由于单烷基取代的草酸脱羧时, 第一次较快, 第二次相对慢, 作者认为提高反应温度对两次脱羧有利. 因此, 较高的温度对提高该反应的产率是有利的.

2019年, Overman等[24a]报道了一例以烷基取代草酸盐62为烷基自由基前体, 实现氮杂芳环的C—H键烷基化反应(图式21a). 该策略需要过量的过硫酸铵作为氧化剂, 进而实现了一系列不同类型杂环, 如: 喹啉、吡啶和喹喔啉等氮杂芳环的烷基化反应. 最近, Yu等[24b]也报道了一例无需额外光敏剂参与的醇与嘌呤、嘌呤核苷环的6号位的脱氧/区域选择性烷基化反应(图式21b). 嘌呤和嘌呤核苷骨架出现在许多生物活性化合物中, 例如, 眩晕药. 因此, 对嘌呤和嘌呤核苷骨架的区域选择性C—H键官能化, 具有重要的研究意义. 在该方法中, 需要添加过量的双三氟乙酸碘苯(PIFA), 而反应成功的关键在于原位生成的烷基取代的草酸与PIFA发生配体交换生成新的高价碘中间体, 该中间体不稳定, 直接经历光敏化, I—O键均裂、脱羧, 产生关键的烷基自由基中间体. 克量级反应和天然活性分子的修饰反应成功展现出该策略的实用性. 例如, 化合物67d在氨水/甲醇体系中脱乙酰基, 得到anti-CEM活性的6-cyclopentyl nebularine (6-CPN) (IC50=16 μmol/L).
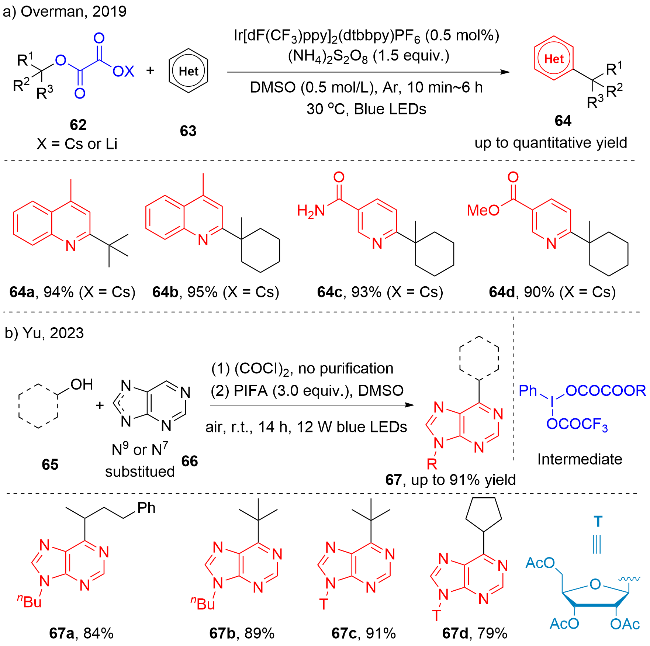
在有机转化中, 炔烃也是一类良好的自由基受体. 2018年, Chu团队[25a]报道了一例可见光驱动烷基取代草酸盐、芳基乙炔和芳基溴代物的三组分的炔烃的烷基化(氮杂)芳基化反应, 合成了一系列三取代的烯烃71(图式22a). 该反应主要涉及烷基自由基对端炔的加成、过渡金属介导的偶联以及烯烃的异构化三个过程, 最终以较高的反应活性和选择性获得多取代顺式烯烃(52%~91% yields). 2020年, 该团队[25b]进一步将该策略拓展至烷基取代草酸盐与烷基取代端炔烃、杂芳基溴代物的三组分的自由基串联反应中(图式22b). 在该反应中主要涉及烷基自由基对端炔的加成、1,5-氢迁移和Ni催化的偶联反应等过程.


对于其反应机理, 作者提出在蓝光的照射下, 三级醇衍生的草酸盐76被激发态的光催化剂*PC单电子氧化, 脱两分子的CO2, 产生三级碳自由基中间体76-1. 接着, 该碳自由基76-1对炔基高价碘试剂77进行自由基加成, 脱邻碘苯甲酸氧自由基, 释放出目标产物二取代的乙炔化合物78. 最后, 邻碘苯甲酸氧自由基与低价态的光催化剂PC•−发生单电子还原, 产生基态的光催化剂PC和邻碘苯甲酸氧负离子, 完成催化循环.
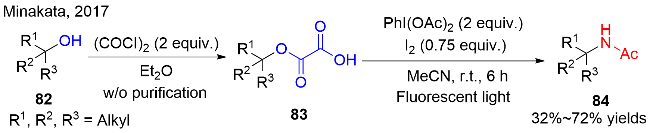

随后, MacMillan等[30c]采用一锅两步的方式发展了一种可见光促进醇类化合物的脱氧/氟化反应(图式25c). 该反应第一步通过生成烷基取代的草酸作为醇的活化试剂; 第二步在金属銥作为光催化剂的存在下, 与Selectfluor反应, 高效率地制备了一系列烷基氟化物93. 与Reisman和Brioche等的工作不同在于, 该反应使用烷基取代的草酸作为醇的活化试剂, Na2HPO4作为无机碱, 丙酮和水作为混合溶剂的条件下, 以相对更高的收率获得目标烷基氟化物. 作者提出: 水在该反应中起到非常重要的角色, 可以增加Selectfluor和Na2HPO4在反应混合液中的溶解度, 提高反应的均匀性, 进而促进反应收率的大幅提高. 该反应针对三级醇和二级醇都能很好的适应, 相对而言, 一级醇脱氧/氟化的效果较差, 可能是一级碳自由基不太稳定的缘由.
关于可见光氧化还原催化醇的脱氧/氟化反应, MacMillan等提出了可能的机理过程, 如图式26所示. 首先, IrIII复合物受可见光激发, 跳跃至其激发态*IrIII (E1/2red [IrIV/*IrIII]=-0.82 V vs. SCE in MeCN), 随后被体系中的少量的Selectfluor (E1/2red=+0.33 V vs. SCE in MeCN)猝灭转化成IrIV复合物. 在无机碱的存在下, 单取代草酸92-1被强氧化性的IrIV复合物氧化、经历两次脱羧过程, 产生烷基自由基中间体92-2. 最后, 该烷基自由基92-2从Selectfluor中攫取氟原子, 得到烷基氟化物93, 同时产生氮自由基阳离子中间体, 进入下一个催化循环. Stern-Volmer荧光猝灭实验证实: 激发态的*IrIII复合物的荧光强度在Selectfluor的存在下可以被降低, 但是化合物92-1活化后的草酸中间体却不能达到这个结果. 因此, 揭示了Selectfluor协助的单电子氧化过程是该光催化反应的初始步骤.

烷基硼酸酯因其C—B键可以较为容易发生多种类型的化学转化, 因而, 在有机合成领域中被视为一类非常有价值的合成子. 在2019年, Studer等[31]报道了一例可见光驱动的烷基取代草酸酯与双联邻苯二酚硼酸酯95的脱氧/硼化反应, 构建了一系列烷基硼酸酯96(图式27). 在温和的反应条件下, 该反应表现出较好的官能团兼容性, 如: 烷基草酸酯底物中包含内烯、苄基胺或酯基片段时, 反应均能顺利发生, 能够以较高收率生成相应硼酸酯. 此外, 作者还发现该自由基硼化策略适用于O-烷基取代的黄原酸酯类衍生物97. 在该反应中, N,N-二甲基乙酰胺作为溶剂时, 提升了底物的溶解度. 三(三甲基硅基)硅烷(TMS)3SiH作为还原剂, 促进O-烷基取代的黄原酸酯类衍生物产生烷基自由基. 值得注意的是: 为避免反应过程中产生的烷基自由基直接被硅烷还原, 需要使用过量的双联邻苯二酚硼酸酯95, 有助于快速捕获烷基自由基, 进而避免副反应的发生.
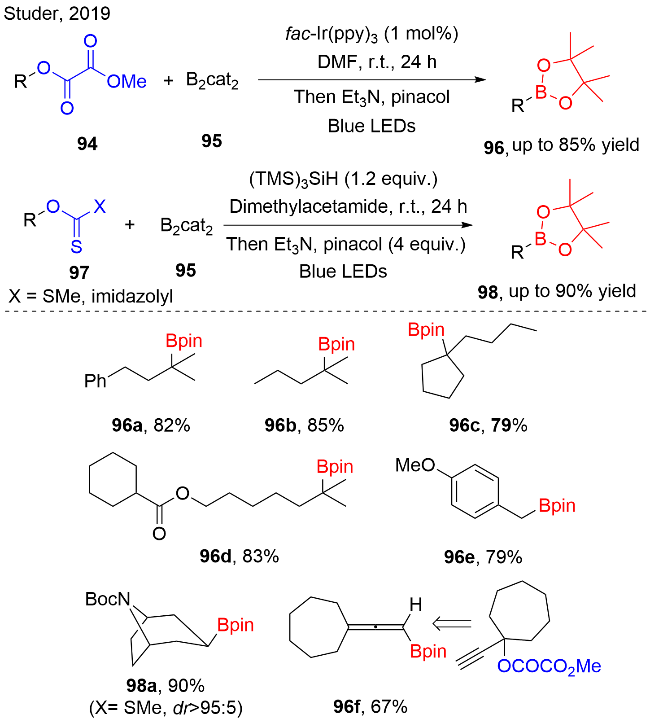
作者根据相关文献, 提出了一个可行的机理途径, 如图式28所示. 首先, 在可见光的照射下, IrIII复合物被激发到其激发态*IrIII. 化合物94被*IrIII复合物单电子还原, 生成自由基负离子中间体94-1, 随后C—O键均裂, 生成烷基自由基. 之后, 烷基自由基被化合物95捕获, DMF作为配体以稳定中间体95-1, B—B键均裂, 释放出烷基硼酸酯96-1, 后续与pinacol作用, 生成目标产物96. 反应中生成的IrIV复合物与硼自由基中间体95-3发生单电子还原, 得到基态的IrIII复合物, 完成催化循环, 同时产生95-4中间体.
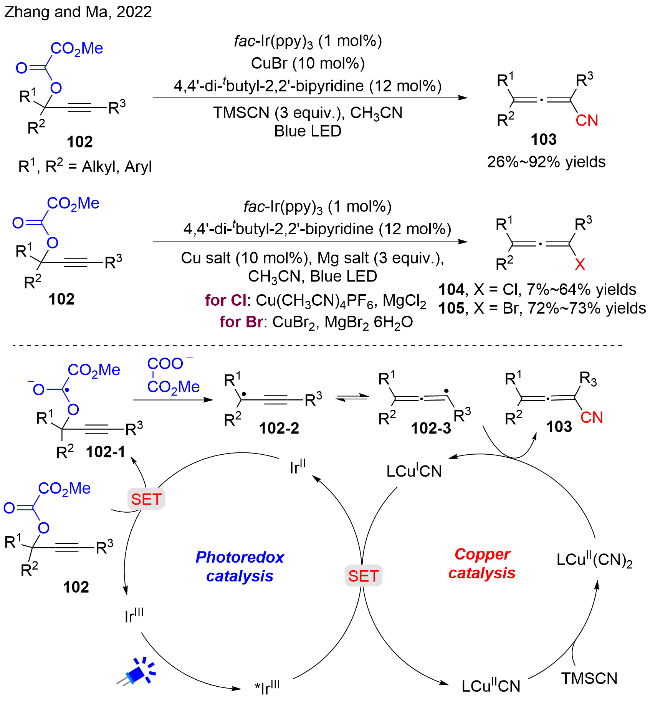
作者通过一系列的机理实验, 如: 荧光猝灭、自由基捕获和控制实验等, 以及DFT理论计算进一步支撑了氧化还原中性的猝灭机理. 其中, Stern-Volmer猝灭实验证实了CuBr和配体的混合物可以高效地猝灭激发态的*IrIII光催化剂. 机理如图式30所示, 首先, 在蓝光的照射下, 激发态IrIII光催化剂与CuI复合物经过还原猝灭过程生成IrII化合物和CuII复合物; 随后, IrII对炔丙基草酸酯102进行单电子还原, 生成自由基阴离子102-1, 并释放基态IrIII催化剂. 自由基阴离子102-1经过C—O键均裂, 产生炔丙基自由基102-2; 而自由基102-2可以互变为联烯基自由基102-3. 该自由基随后被原位生成的Cu(CN)2复合物捕获, 再经过还原消除过程生成目标产物103, 并释放CuI复合物, 完成催化循环.
4 硫代碳酸酯及其衍生物的脱氧/功能化反应
Barton-McCombie 脱氧反应是有机合成转化中的重要工具之一, 但是在方法学上也遭受到一定的限制, 目前, 一些研究团队持续对其进行改进. 例如: 2014年, Ollivier等[34]利用醇与硫代碳基二咪唑反应一步制备了O-硫代氨基甲酸酯类化合物107, 以其作为醇的活化试剂, 在光氧化还原催化下, 实现醇的脱氧/还原反应(图式31). 该反应以fac-Ir(ppy)3为光催化剂, Hünig’s base 作为电子供体, 在温和的反应条件下, 实现二级和三级醇的脱氧/还原反应, 并以较好的收率获得还原产物108, 包括复杂结构的醇类化合物都能很好的被适用. 该反应的优点在于避免了Barton-McCombie 脱氧反应中对有毒锡化合物的使用.
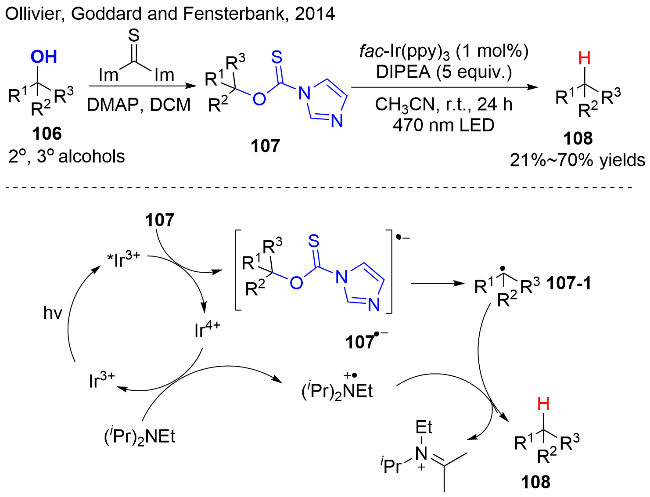
反应的主要机理途径如图式31所示: 首先, 在可见光的照射下, IrIII达到其激发态*IrIII. 接着, 作者通过荧光猝灭实验证实, *IrIII与O-硫代氨基甲酸酯107之间发生单电子还原, 可以获得IrIV和O-硫代氨基甲酸酯的自由基阴离子物种, 其经C—O键均裂得到烷基自由基中间体107-1. 同时, IrIV被三级胺还原至其基态IrIII, 同时产生三级胺自由基阳离子物种. 最后, 烷基自由基107-1在三级胺或其自由基阳离子的存在下, 夺氢, 生成氢化产物108. 在该反应中, 三级胺DIPEA发挥着重要的作用, 不仅作为电子供体, 还作为氢原子供体, 促进还原产物的生成.
2017年, Molander等[35]成功发展了以苄醇衍生的黄原酸酯为底物, 光氧化还原催化和镍催化协同促进C(sp3)—C(sp2)的偶联反应. 在该反应中, 作者通过光催化生成的仲丁基自由基作为活化试剂, 促进O-苄基黄原酸酯化合物中的C—O键断裂, 从而产生苄基自由基中间体. 之后进入镍催化循环, 苄基自由基被零价镍复合物捕获, 产生一价镍复合物, 接着与芳基溴化物发生氧化加成, 得到三价的镍复合物. 三价镍经历还原消除, 释放出偶联产物111, 同时产生一价镍, 再被低价态的光催化剂还原, 回到零价镍物种, 完成催化循环, 如图式32所示. 控制实验暗示无仲丁基三氟化硼钾盐110时, 检测不到目标产物; 当O-苄基黄原酸酯化合物109缺失时, 作者分离出了仲丁基与芳基的偶联产物112; 副产物烷基二硫代碳酸酯109-1的产生, 也证实了仲丁基自由基的存在和活化作用机制.
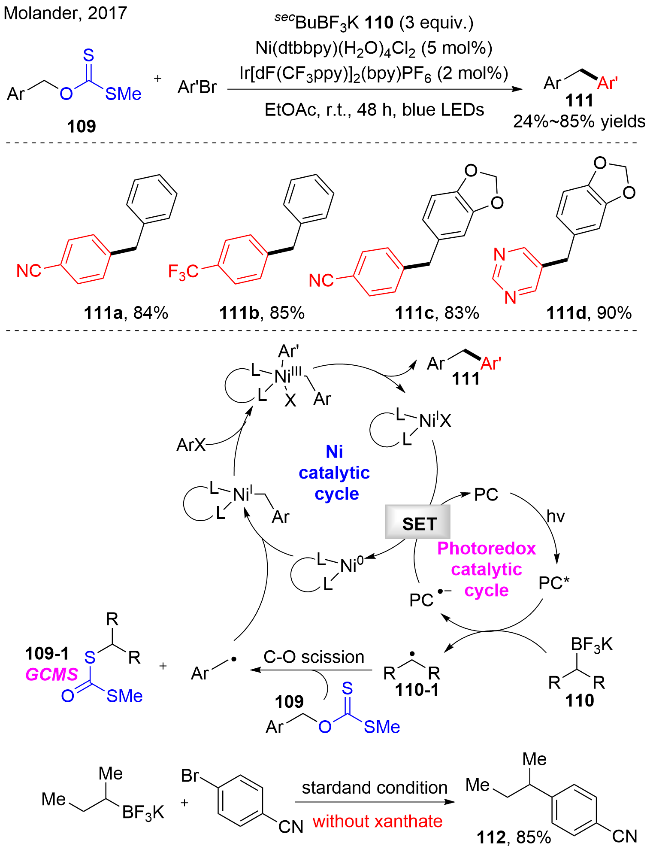
在合成化学中, 烷基硼化合物是一类非常有用的试剂, 例如, 可以作为偶联试剂. 一些结构稳定的烷基硼化合物, 例如: 硼酸酯和三氟硼酸钾盐, 被广泛地用于有机合成、医药化学和材料科学领域. 因此, 对烷基硼化合物的制备是有重要的研究价值的. 2019年, Aggarwal等[36]成功开发了可见光诱导脂肪醇衍生物的脱氧/硼化反应(图式33). 该反应是以O-2-碘芳基-硫代碳酸酯113为脂肪醇的预先活化试剂, 在无光催化剂的存在下, 实现Barton-McCombie 脱氧反应. 该反应的底物范围较为广泛, 其中二级碳自由基, 相较于一级和三级碳自由基, 可以提供较高产率的脱氧/硼化产物116. 同时, 该策略还可以适用于复杂结构的天然产物, 例如menthol, Corey lactone, thymidin, rockogenin, glycyrrhetinic acid等, 都可以较好地实现脱氧/硼化.

控制实验证实三乙胺和光源对该反应非常重要, 无光条件下, 无硼化产物的生成; 无三乙胺时, 反应的效率大大降低; 自由基钟实验说明有脱氧烷基自由基的存在; 因此, 作者提出了可能的机理历程, 如图式34所示. 首先, 双联邻苯二酚硼酸酯114 (B2cat2)与三乙胺形成路易斯酸-碱加成物, 随后与硫代碳酸酯113, 在可见光的驱动下, 形成EDA复合物, 通过紫外可见光谱实验得到证实. 此外, 实验证明碘原子在Barton-McCombie 脱氧反应中起到非常重要的作用, 一方面: 反应过程中环状的硫代碳酸酯113-2的生成, 说明EDA复合物中C—I键被还原, 有芳基自由基113-1的存在; 另一方面: 碘负离子作为配体可以增加硼原子上邻苯二酚的电子云密度. 接着, 113-1发生分子内自由基环加成, 生成中间体113-2, 随后C—O键均裂, 产生烷基自由基. 之后, 烷基碳自由基被B2cat2捕获, 释放出烷基硼酸酯115和硼自由基阴离子中间体Int-1. 最后, 该硼自由基阴离子Int-1被113通过单电子转移过程猝灭.

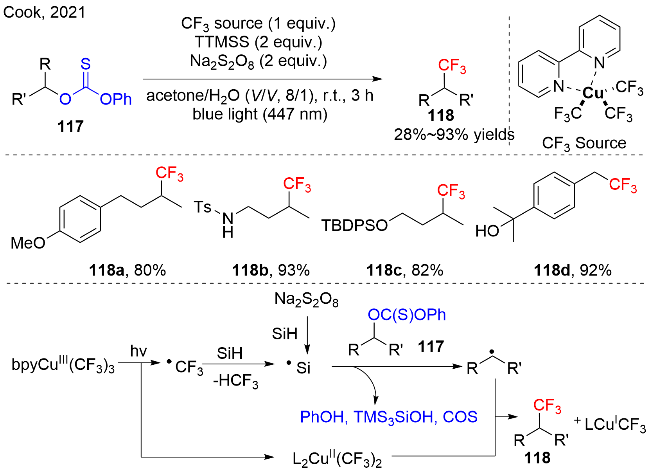
作者根据相关文献和机理实验以及控制实验证明, 在光照条件下, 铜-三氟甲基复合物可以产生三氟甲基自由基和二价铜复合物. 在反应过程中, 硅基自由基可以通过Na2S2O8氧化硅烷快速产生, 同时也可以通过三氟甲基自由基协助硅烷产生. 接着, 硅基自由基与O-烷基-硫代碳酸酯117作用, 生成烷基自由基, 释放出副产物苯酚、硅醇和氧硫化碳. 最后, 烷基自由基被二价铜-三氟甲基复合物捕获, 产生三氟甲基化产物118和一价铜复合物, 如图式35所示. 作者证实无光源或无硅烷, 没有三氟甲基化产物; 无氧化剂或者无水或者在空气中进行, 该反应的产率很低, 暗示这些因素都对该反应的成功起到非常重要的作用, 都是不可缺少的.
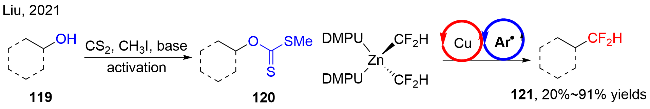
2022年, Hong等[39]发展了“一锅两步”的策略, 可见光直接促进醇或者硫醇的脱氧(脱硫)/区域选择性吡啶化反应(图式37). 该策略以黄原酸盐123为醇(硫醇)的活化试剂, 在有机三价磷的存在下, 无光催化剂存在下, 与N-氨基吡啶盐124反应, 实现吡啶位点选择性的烷基化反应. 在该反应中, 有机磷对反应的成功起到非常重要的作用; 同时, 可见光对该反应的效率起到一定的促进作用, 即在黑暗条件下, 产率稍微有所下降. 该反应展示了良好的官能团兼容性, 底物适用性非常广泛, 包括伯醇、仲醇和叔醇, 以及各种硫醇化合物, 都能较好地实现脱氧(脱硫)过程. 当底物122中含有二醇结构时, 反应对于位阻小的的二级醇选择性比三级醇更高. 此外, 对一些天然产物和生物活性分子, 以及复杂结构的化合物, 也能很好地进行脱氧反应.
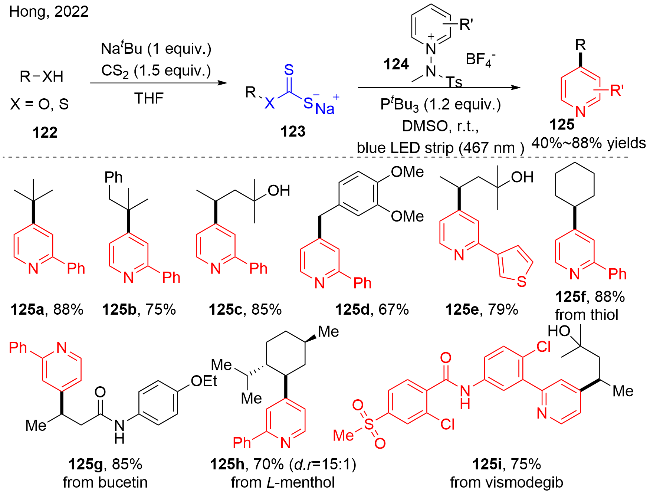
对于该反应的可能性机理, 作者通过机理实验和理论计算提出, 如图式38所示: N-氨基吡啶盐与三叔丁基膦之间形成对抗的路易斯酸-碱对(FLP), 通过紫外可见光谱实验证实峰位置发生偏移. 随后, FLP之间的单电子转移过程致使产生膦自由基阳离子物种. 膦自由基阳离子与黄原酸盐123作用, 生成烷基自由基, 同时释放出CXS和硫膦化合物. 接着, 烷基自由基对N-氨基吡啶盐124进行自由基加成, 生成烷基取代的吡啶产物125和氮中心自由基. 作者提出氮中心自由基的产生有助于发生自由基链反应过程, 却未通过量子产率进行证实. 此外, 作者通过紫外可见光谱实验揭示: 黄原酸盐与N-氨基吡啶盐之间存在相互作用, 该光反应过程可以产生黄原酸硫自由基123-1, 参与自由基链反应, 能够稍微提高反应的产率.

2021年, Wu等[40]报道了可见光氧化还原催化脂肪醇的脱氧/ Giese反应(图式39). 该反应采用O-烷基黄原酸盐127作为醇的活化前体, 三苯基膦作为有效的氧原子转移试剂促进O-烷基黄原酸盐的脱氧, 生成活性的烷基自由基物种. 该反应在温和反应条件下, 具有广泛的底物范围, 适用于伯、仲、叔醇的底物, 以及二醇的选择性脱氧化反应. 同时, 该策略通过“一锅两步”的方式也成功地实现了药物分子和天然产物分子的脱氧/烷基化反应. 该反应将对天然产物全合成中特别是分子内环化反应中的作用巨大. 控制实验揭示: 无光, 或者无光催化剂, 或者无三苯基膦, 反应都没有目标产物的生成; 在无水条件下, 产率会大大降低, 原因是: 水一方面作为质子源, 另一方面可以提高黄原酸盐在体系中的溶解度, 进而提高反应的效率.
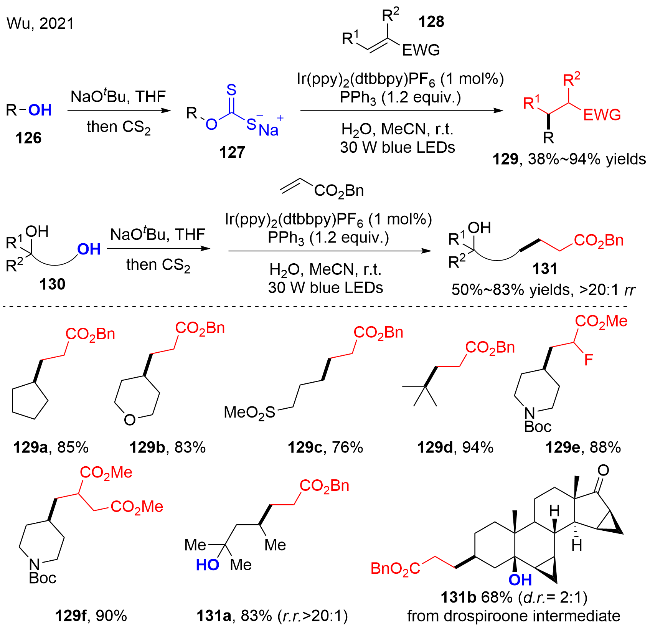
作者通过TEMPO和自由基钟实验证实了烷基自由基的存在; 另一方面Stern-Volmer荧光猝灭实验说明O-烷基黄原酸盐能够更加高效地猝灭激发态的*IrIII光催化剂. 机理途径如图式40所示, 在光氧化还原催化的条件下, O-烷基黄原酸盐127发生单电子氧化, 产生O-烷基黄原酸硫自由基物种127-1. 接着, 三苯基膦与硫自由基物种127-1作用, 生成膦自由基物种127-2, 随后通过C—S键和C—O键的均裂, 释放出硫化膦化合物和氧硫化碳, 以及生成关键的烷基自由基. 之后, 烷基自由基对缺电子烯烃128进行自由基加成反应, 还原, 夺氢, 获得烷基化产物129.
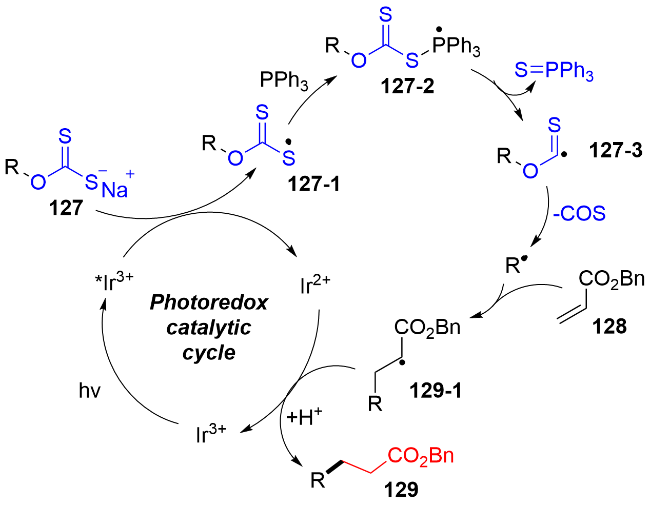
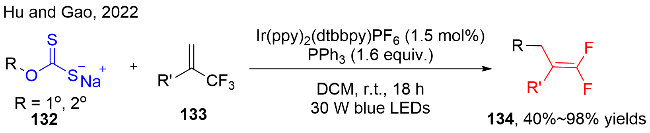
之后, Wu课题组一直致力于研究醇衍生的黄原酸盐的脱氧/功能化反应. 与之前可见光氧化还原催化策略[40]不同, 该课题组也成功发展了在无光催化剂的条件下, 在有机膦的存在下, 可见光促进黄原酸盐与缺电子结构化合物的脱氧/功能化反应. 例如: O-烷基黄原酸盐分别与烯基砜[42a]136、N-苄氧基磺酰基亚胺[42c]140、杂芳环砜类化合物[42d]142、对苯二腈反应[42e]144, 可以获得烷基取代的烯烃137(图式42a)、亚胺141(图式42c)、杂芳烃143(图式42d)和芳烃145(图式42e); 此外, 与N-磺酰基亚胺138发生自由基加成反应[42b], 可以获得磺酰胺类化合物139(图式42b). 在2023年, Zhang和Han团队[42f]采用类似的策略开发了可见光直接促进醇衍生的黄原酸盐的脱氧/二氟乙烯化反应(图式42f).
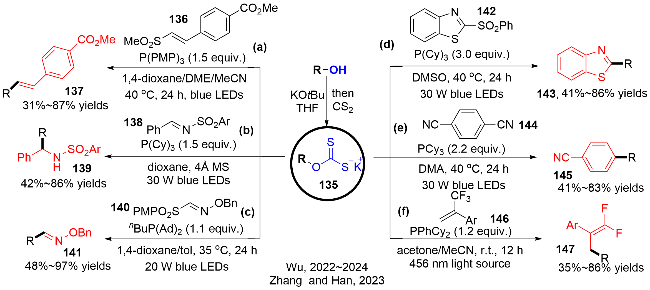
以N-磺酰基亚胺138为例, 其反应机理如图式43所示. 作者通过黄原酸盐135与N-磺酰基亚胺混合物的紫外可见光谱实验证实: 反应过程中不存在EDA复合物的生成. Stern-Volmer荧光猝灭实验揭示了黄原酸盐135可以被N-磺酰基亚胺猝灭. 反应首先在可见光的照射下, 黄原酸盐135转变成其激发态(*E1/2=-2.4 V vs. SCE in MeCN), 随后与N-磺酰基亚胺138 (E1/2=-1.45 V vs. SCE in MeCN)发生单电子转移过程, 该过程也是热力学上支持的, 生成硫自由基物种135-1和亚胺自由基阴离子物种138-1. 之后, 硫中心自由基与三环己基膦作用, 发生C—S键和C—O键的均裂, 释放出硫化膦化合物和氧硫化碳(COS), 同时生成关键的烷基自由基活性中间体. 最后, 烷基自由基与亚胺自由基阴离子138-1发生自由基-自由基偶联反应, 质子化, 获得磺酰胺产物139. 此外, 反应呈现的低的量子收率(Φ=0.022)证实了该反应不涉及自由基链反应的过程.
5 醚类化合物的脱氧/功能化反应
醚类化合物也是一种常见的, 容易获取的, 无毒的含氧化合物. 在过去的几十年里, 醚类化合物, 尤其是芳醚和乙烯基醚, 通常是通过金属催化醚键C—O键的断裂参与的偶联反应, 实现醚类的脱氧/功能化[43]. 接下来我们将介绍由醇衍生的醚类化合物在光化学反应中发生的脱氧/官能化反应.
醛作为有机合成中一种容易发生多样化反应的官能团. 通常, 多取代的烷基醛需要通过多步官能团转化才能获得. 在2018年, Huang, Nicewicz 和Chen等[44]报道了一种一步法由容易获取的烯烃和乙烯基醚, 在光氧化还原催化的条件下, 合成了多取代的烷基醛的方法(图式44). 该方法以吖啶盐作为有机光催化剂, 二苯基二硫醚作为氢原子转移试剂. 该反应具有宽广的底物范围, 以优异的化学选择性和区域选择性, 中等到优秀的收率可以制备α-, α,β-, β,γ-和α,β-,γ-支链的烷基醛化合物150. 此外, 一些天然产物衍生的烯烃也能较好地应用到该反应中, 例如, estrone, diacetone-D-glucose和totarol的类似物. 利用这种方法可以拓展生物活性分子的衍生物.
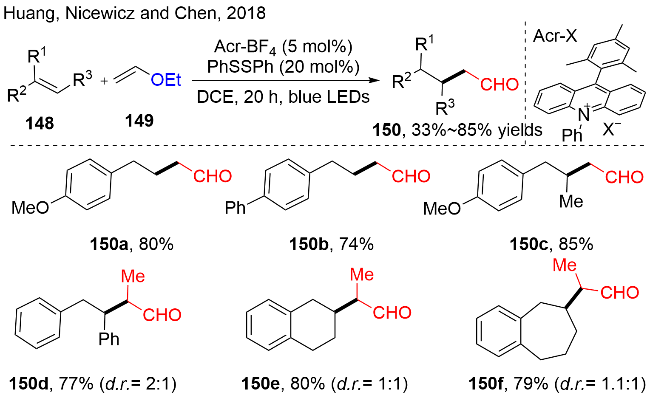
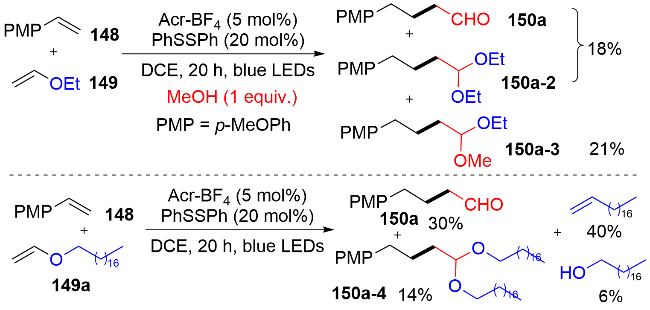
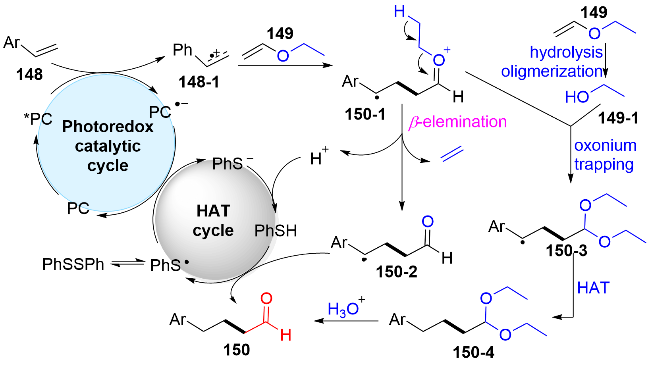
对于其反应机理, 作者通过在模版反应中加入甲醇或者使用长脂肪链的乙烯基醚, 检测到反应中有不同结构缩醛和脂肪链烯烃的产生(图式45). 这些机理实验说明反应可以通过多种途径得到目标产物. 因此, 作者提出机理如图式46所示: 首先由于良好的π-π堆叠, 激发态的吖啶盐*PC (E*1/2(PC*/PC•−)=+2.06 V)更倾向于氧化富电子的苯乙烯(+1.15 to +1.75 V vs. SCE), 从而得到苯乙烯自由基阳离子中间体148-1. 随后, 与乙烯基醚149反应, 得到关键中间体150-1. 第一种途径, 该中间体150-1发生β-C—H键断裂, 释放出烯烃, 同时生成苄基自由基结构的醛150-2. 另一种途径, 中间体150-1中的氧鎓盐结构与乙烯基醚水解后产生的乙醇发生亲核加成, 生成连有缩醛结构的苄基自由基中间体150-3. 在氢原子转移(HAT)循环中, 二苯基二硫醚均裂产生的苯硫基自由基, 与还原性的PC•−发生单电子转移, 得到基态PC和苯硫负离子, 随后质子化产生苯硫酚. 最后, 中间体150-2和150-4分别经历攫氢和酸化水解, 产生目标产物烷基醛150.
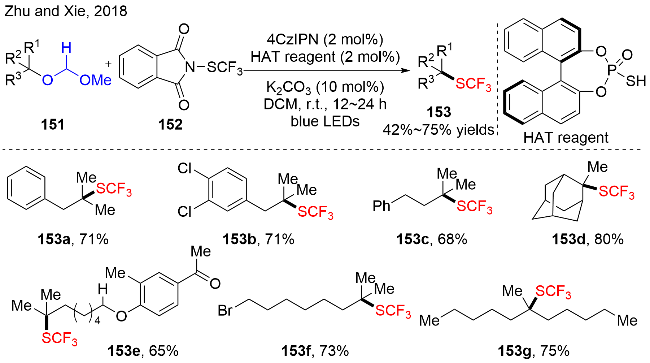
反应机理如图式48所示, 作者提出: 在光氧化还原催化下, 硫负离子被激发态的光催化剂氧化为硫中心自由基. 随后, 在HAT循环中, 硫自由基选择性地夺取MOM类化合物151中的H1原子, 生成亚甲基碳自由基151-1和硫醇. 接着, 亚甲基自由基151-1发生邻位的 C—O键断裂, 释放出副产物甲酸甲酯和高活性的三级烷基碳自由基151-2中间体. 最后, 三级碳自由基151-2与N-硫三氟甲基取代的邻苯二甲酰亚胺(Phth) 152作用, 获得硫三氟甲基化产物153和邻苯二甲酰亚胺氮中心自由基. 该氮中心自由基被低价态的PC•−还原, 释放出邻苯二甲酰亚胺氮负离子和基态的PC.

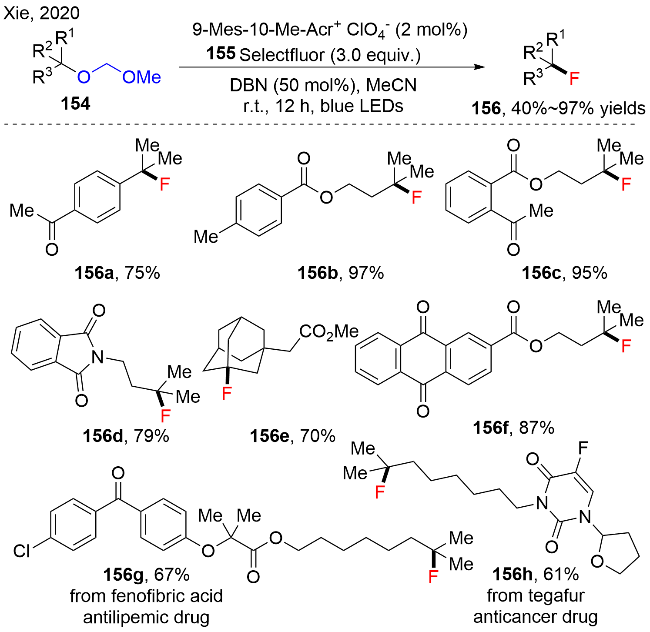
机理如图式50所示, 作者通过荧光猝灭实验, 证实1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)作为还原剂能更加容易猝灭激发态的光催化剂PC*, 产生低价态的光催化剂PC•−和三级胺自由基阳离子. 随后, 在HAT循环中, 三级胺自由基阳离子中间体与MOM三级醚154发生HAT过程, 释放出甲酸甲酯, 它可以通过1H NMR检测到, 以及生成烷基碳自由基154-2. 之后, 烷基自由基154-2与Selectfluor 155反应, 生成氟化产物156, 同时生成中间体155-1. 中间体155-1被低价态的光催化剂PC•−还原, 结束光催化循环. 此外, 作者通过测定该反应的量子产率(Φ=0.7), 以及用15%的偶氮二异丁腈(AIBN)代替光催化剂进行反应, 结果显示仍能以16%的产率得到目标产物. 这些实验证实了反应中可能存在自由基链传递的过程. 自由基链传递途径为: 三级MOM醚154也可以与中间体155-1发生HAT过程, 生成烷基碳自由基154-2, 攫取氟, 生成目标产物.
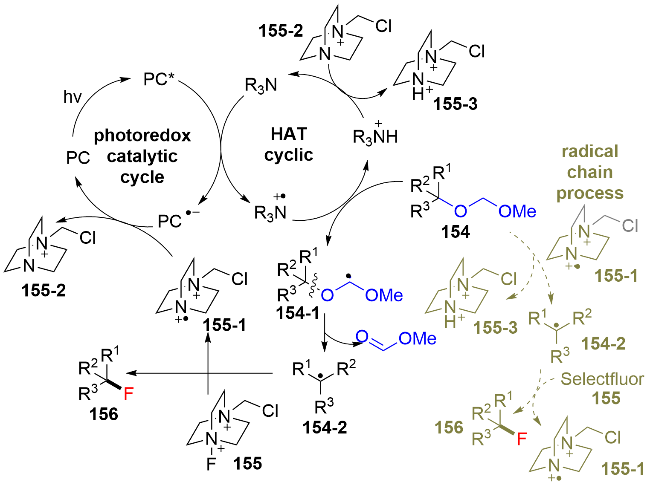
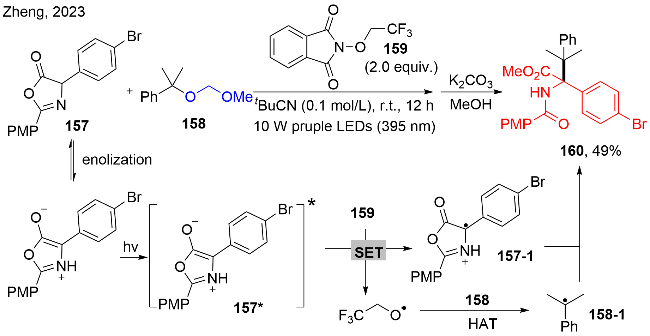
6 其他醇衍生物的脱氧/功能化反应
除了上述常见的醇的含氧衍生物, 还有一些氮氧化合物, 可以由醇衍生得到. 例如: N-烷氧基邻苯二甲酰亚胺[48], 通常是作为烷氧基自由基前体, 再经历氢原子转移或者β-键断裂, 诱导新的碳自由基产生.
2017年, Chen等[48b]发展了可见光诱导烷氧基自由基的产生, 并导致的选择性C(sp3)—C(sp3)键的断裂反应(图式52). 其中, 光源和汉斯酯HE对该反应的成功起到非常重要的作用. 作者通过161和HE混合物的紫外可见光谱实验, NMR滴定实验确定了它们之间存在相互作用, 即有它们的EDA复合物的生成. 作者还证明蓝光相对于短波长范围的450 nm和475 nm, 更能促进EDA复合物的形成. 该反应通过形成161和HE的EDA复合物, 产生烷氧基自由基. 随后, 烷氧基自由基发生β位C(sp3)—C(sp3)键的断裂, 释放出简单醛和烷基自由基R1. 最后与烯丙基苯亚磺酸酯162反应, 获得烯丙基化的产物163. 该反应底物反应宽广, 无论是伯醇、仲醇还是叔醇衍生的氮氧化合物, 都能较好地参与反应. 此外, 氧原子β位C(sp3)—C(sp3)键断裂后, 新形成的烷基自由基, 无论是一级和二级, 还是大位阻的三级烷基碳自由基, 也都能很好地与162进行自由基加成反应. 与此同时, 作者还尝试了与苯乙烯基苯亚磺酸酯的反应, 得到了烯基化的产物163h. 该反应的一个小缺点是对HE的用量较大, 原子经济性较差.

2020年, Martin等[49]利用类似的策略产生烷基自由基, 在光氧化还原催化与镍协同催化下实现C(sp3)—C(sp2)和C(sp3)—C(sp3)键的偶联反应(图式53). 需要注意的是: 该反应在无镍复合物, 或者无HE或者无光照条件下, 都不会发生反应; 但无光催化剂时, 可以获得63%核磁收率(标准条件下84%核磁收率). 这些控制实验说明HE和光源对该反应的成功起到非常重要的作用. 在底物的适用性方面, 该反应对一级醇、二级醇和三级醇衍生的氮氧化合物164, 其中涉及一级、二级和三级烷基碳自由基, 都能以41%到97%的分离收率获得目标产物165. 该反应的官能团兼容性也很好, 例如: 甲硫基、未保护的二级胺、磺酰胺、硼酸酯以及酯基都能够兼容. 除此之外, 作者还将该策略拓展到醇的含氧衍生物与烷基溴化合物的C(sp3)—C(sp3)键偶联反应中, 并取得中等到良好的收率. 该方法也能对一些复杂结构的药物分子、氨基酸和生物活性分子, 都能成功地进行结构修饰, 例如celecoxib、mannopyranose和galactopyranose进行嫁接或者开环芳基化修饰(图式54).

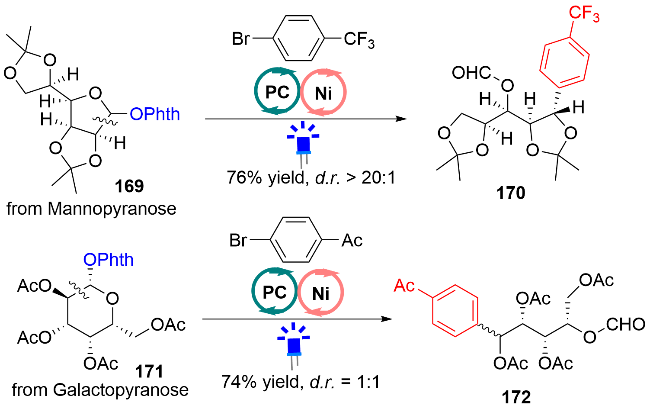
对于该反应的机理, 作者通过氮氧化合物164和HE的紫外可见光谱实验证实, 吸收峰发生红移, 它们之间形成了EDA复合物. 氮氧化合物164和HE的EDA复合物的解离常数KEDA也被计算出来, 为2.9 L/mol. TEMPO-烷基自由基的加成物也被MS捕获到. 此外, 作者还制备出Ni-I复合物, 并将其应用在C(sp3)—C(sp2)偶联反应中, 成功地获得了目标产物. 为此, 作者提出了光氧化还原催化与镍协同催化C—C键偶联反应的机理, 如图式55所示. 氮氧化合物164和HE的EDA复合物, 在蓝光的照射下, 发生单电子转移, 得到164-1物种, 经N—O键断裂, 产生烷氧基自由基164-2, 随后发生β位C(sp3)—C(sp3)键均裂, 释放出醛, 生成关键的烷基自由基164-3. 接着, 该烷基自由基被氧化加成后的NiII复合物捕获, 后经还原消除, 释放出偶联产物和NiI复合物. 最后, NiI复合物被低价态的光催化剂PC•−还原, 重新产生零价镍和基态的光催化剂PC, 完成双催化循环.

2021年, Cai等[50]报道了可见光促进N-烷氧基邻苯二甲酰亚胺通过β-断裂产生烷基自由基, 与甘氨酸衍生物的C(sp3)—C(sp3)键偶联反应(图式56). 该方法操作简单, 反应条件较温和, 能够以23%~92%的收率获得一系列非天然的复杂结构的氨基酸和多肽类化合物. 同时, 该方法还可以成功地应用于saclareolide, β-pinene和camphor的修饰, 直接与甘氨酸衍生物相连接. 该方法具有底物范围广, 官能团兼容性好, 例如, 在标准反应中加入生物活性分子, 有些能够促进反应产率的提高等特点. 通过活性检测, 有些修饰的氨基酸和多肽类化合物具有体外抗真菌的活性特征. 作者提出该反应是通过烷基自由基与α-氨基自由基交叉偶联的途径实现的.

由醇衍生的氮氧化合物, 除了上述烷氧基自由基促进的β位C(sp3)—C(sp3)键的断裂, 释放简单醛或者酮, 生成烷基自由基进行的功能化反应之外, 烷氧基自由基也可以被三价膦捕获, 促进其α位C—O键的断裂, 产生新的碳自由基. 类似的, 在有机膦化合物的存在下, 通过自由基的途径, 也可以实现醇类化合物的直接脱氧转化, 已经有相关综述对其进行详细介绍, 在此我们不再进行赘述[8].

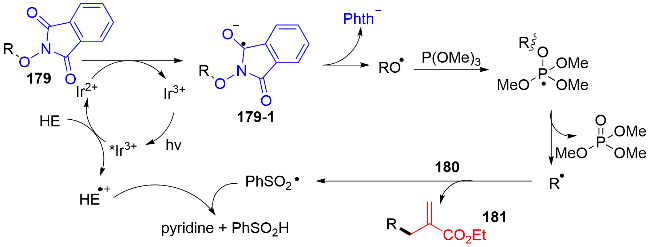
作者通过Stern-Volmer荧光猝灭实验证实: 汉斯酯可以成功地猝灭激发态的金属铱催化剂, 而三甲氧基膦不能. 因此, 可行性的机理途径如图式58所示. 在可见光的照射下, 激发态的PC*与HE发生单电子还原猝灭, 生成IrII和HE自由基阳离子物种. 随后, IrII与氮氧化合物179发生单电子转移, 得到基态的IrIII和自由基阴离子物种179-1. 该179-1物种经历N—O键均裂, 产生烷氧基自由基. 之后, 烷氧基自由基被三甲氧基膦捕获, 脱去氧化膦, 获得关键的烷基自由基中间体. 最后, 烷基自由基对烯丙基砜180进行自由基加成, 得到烯丙基化产物181. 苯亚磺酰基自由基与HE的自由基阳离子作用, 产生苯亚磺酸和HE的吡啶副产物, 结束反应循环.
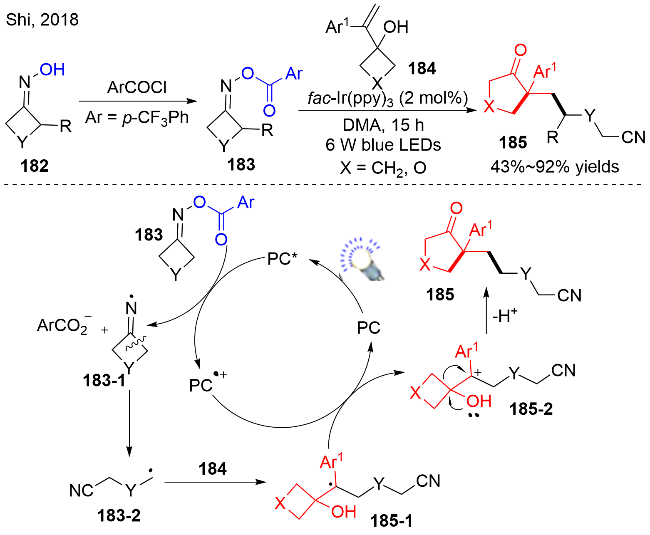
除了醇衍生的氮氧化合物, 一些其他的含羟基结构的化合物, 例如N-羟基亚胺化合物, 也能发生类似醇羟基脱氧的反应. 2018年, Shi等[52]以N-羟基亚胺化合物182为底物, 酯化得到中间体183, 之后在可见光氧化还原催化的条件下, 与环烷基取代苯乙烯化合物184进行多米诺反应, 得到N-羟基亚胺化合物脱羟基功能化的产物185. 反应机理如图式59所示, 首先, 在可见光的照射下, 激发态的光催化剂PC*与底物183发生单电子转移, 底物183被还原发生C—O键断裂, 得到N中心自由基中间体183-1, 同时得到氧化态的光催化剂PC•+. 中间体183-1发生开环, 产生含氰基结构的烷基自由基183-2, 随后与底物184发生自由基加成, 氧化得到苄基碳正离子中间体185-2. 最后, 中间体185-2发生Sem-pinanol重排, 脱质子, 获得产物185.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
7 总结与展望
醇类化合物来源丰富、便宜易得. 醇的脱氧/功能化反应被认为是一种实现碳链重塑和官能团转化的有效策略之一. 对于醇羟基的直接脱氧化的催化模式, 之前已经有文献和综述报道过. 但是, 醇的直接脱氧/功能化反应, 也有不足之处, 例如, 需要预活化羟基或者先转化成其他易离去的官能团之后, 才能够发生取代反应; 在这种反应条件下, 反应类型单一, 限制了醇的合成应用前景. 此外, 借助光化学或者电化学催化的条件下, 过量的三价膦化合物也可以促进醇的直接脱氧转化, 被认为是一种非常重要的合成转化方法. 但是, 这种双重催化产生自由基的策略, 一般会使反应类型和底物的普适性受到一定的局限, 例如: 三级醇比一级和二级醇效果表现的更好. 此外, 对于醇的直接脱氧化/功能化反应在不对称合成领域的应用, 文献报道的较少.
在这篇综述中, 我们着重介绍近十年来不同类型的醇的含氧衍生物在光化学反应的条件下, 进行脱氧化/功能化反应的研究进展, 包括羧酸酯、草酸酯、硫代碳酸酯及其衍生物, 醚类化合物和其他类型的含氧化合物. 光化学催化醇的含氧衍生物的脱氧的途径, 主要取决于醇的衍生物的结构, 可以通过单电子转移或者氢原子转移的方式, 促进醇的衍生物中C—O键的断裂, 发生脱氧过程, 进而生成碳自由基中间体; 或者是通过单电子转移的途径生成的氧中心自由基, 与有机膦化合物作用, 释放出膦氧化合物, 从而实现醇的脱氧, 后续进行功能化反应, 最终实现碳链的重塑或者官能团的转化. 在这里, 我们具体地介绍在可见光的驱动下, 不同结构类型的醇的含氧衍生物的催化脱氧的模式、底物的普适性、反应机理和反应优缺点.
另外, 该综述将对其他的含羟基(例如: 羧酸, 羟胺, N-羟基亚胺等)[53]或含氧结构化合物的脱氧化/功能化反应的实现, 具有一定的指导意义. 最近, 我们团队也正在进行有关醇的含氧衍生物的脱氧化反应的相关工作. 在化学合成领域中, 将来可能会有越来越多的研究人员使用生物质衍生的碳源来代替化石碳源, 并可能会对催化去功能化方法产生强烈的需求.
(Cheng, B.)