

1 引言
烯烃的选择性氧化裂解反应是有机合成中常用来引入含氧官能团的反应之一, 其产物芳香酮类化合物在医药、香料、染料、食品等领域都发挥着十分重要的作用[1]. 目前, 传统的合成方法包括臭氧分解反应[2]或Lemieux-Johnson氧化反应[3], 但安全问题和昂贵的试剂使它们仍无法满足绿色化学和有机合成中原子经济性的要求. 常用的替代方法是使用过渡金属催化(如Ru[4], Pd[5], Au[6], Pt[7], Fe[8]和Cu[9])或热引发的自由基过程[10], 并且结合各种氧化剂进行的热化学反应, 例如过硫酸氢钾(Oxone), 间氯过氧苯甲酸(mCPBA), PhI(OAc)2, 叔丁基过氧化氢(TBHP), NaIO4和H2O2(图1, Eq. (a)). 尽管转化相对的简单, 但这些方法需要使用昂贵的金属催化剂、化学计量的氧化剂以及添加剂, 通常会导致有毒副产物的产生, 不符合绿色化学的要求. 因此, 如何实现温和、高效且可持续的烯烃氧化裂解反应, 具有重要的理论意义和现实要求. 相较于以上的氧化剂, 氧气是最安全、丰富易得的氧化剂, 避免了有毒和化学计量的副产物, 在烯烃氧化中的应用长期以来受到人们的广泛关注.

近年来, 光催化反应领域的兴起为有机合成开辟了一条绿色的新途径[11]. 可见光作为清洁、可再生的能源被广泛应用于促进各种有机合成反应, 特别是利用分子氧和有机小分子光催化剂发展的几种烯烃C=C双键的氧化裂解体系[12]. 在激发态光催化剂的作用下, 烯烃被光氧化生成相应的自由基阳离子, 然后与不同形态的分子氧发生光催化反应. 分子氧可表现出三种氧化态: 分子氧(O2)、单线态氧(1O2)和超氧自由基阴离子($O_{2}^{\centerdot -}$), 其中与分子氧作用的光催化剂是: 10-甲基吖啶高氯酸盐($Acr{{H}^{+}}ClO_{4}^{-}$)[13], 与单线态氧作用的光催化剂是: 5,10,15-三苯基-20-(4-羟基苯基)21H,23H-卟啉(TPP-OH)[14]; 与超氧自由基阴离子作用的光催化剂是: 苯亚磺酸钠盐[15a], 聚合氮化碳(PCN)[15b], 9,10-二氰基蒽(DCA)[15c,15d], 二甲氧基苯[15e], 溶剂红(Eosin Y)[15f], 酸性红94[15g](图1, Eq. (b)). 此外, 烯烃的氧化反应也可通过光引发的自由基加成过程发生, 例如, 可见光介导的碘单质[16]和富电子芳香二硫化物[17]催化体系. 然而, 在有机小分子光催化剂的体系中, 若想顺利产生活性氧物种, 则需加入氧化剂、添加剂或碱以确保适度的产率. 同时, 还存在选择性较低, 副产物多等缺点, 不符合绿色化学的基本原则. 因此, 开发一种环境友好、高效且操作简便的烯烃C=C双键氧化裂解的新方法已成为当前研究的热点之一.
2 结果与讨论
2.1 反应条件优化
在30 W蓝光LED灯(400~450 nm)的照射下, 以1,1-二苯乙烯(S1)作底物, 室温条件下初步探究了不同类型的有机小分子光催化剂, 结果如表1所示. 当使用10-甲基-9-均三甲苯基吖啶高氯酸盐([Mes-Acr+- Me$ClO_{4}^{-}$])、溴化乙啶、2,4,6-三苯基四氟硼酸盐和有机染料光催化剂伊红B (Eosin B)、伊红YS (Eosin YS)、罗丹明B (Rhodamine B)时, 仅达到一般的反应效果(表1, Entries 1~6). 4-甲苯硫酚的催化性能优于其它有机小分子光催化剂, 以46%的收率获得了二苯甲酮1(表1, Entry 7). 此外, 用其它溶剂替代乙腈, 例如, 水, N,N-二甲基甲酰胺(DMAc), 二甲基亚砜(DMSO), 1,2-二氯乙烷(DCE), 四氢呋喃(THF), 1,4-二氧六烷, 乙醇(EtOH), 异丙醇(IPA)和甲醇(MeOH)(表1, Entries 8~16), 实验结果表明, 该反应在甲醇中有很好的转化, 二苯甲酮1的产率提高至63%, 表明溶剂对这一转化有较大影响(Entry 16). 最后, 通过改变反应氛围发现高的氧气浓度加速了反应的进行, 二苯甲酮1的收率提升至85%(表1, Entry 17). 综上, 最优的反应条件如下: 以S1作底物, 20 mol%的4-甲苯硫酚作光催化剂, 氧气作氧化剂, 甲醇作溶剂, 在30 W蓝光LED灯的照射下, 室温反应12 h, 以85%的收率获得二苯甲酮1.
表1 实验条件的优化Table 1 Optimization of reaction conditionsa |

| Entry | Photocatalyst | Solvent | Yieldb/% |
|---|---|---|---|
| 1 | [Mes-Acr+-Me$ClO_{4}^{-}$] | CH3CN | 20 |
| 2 | Ethidium bromide | CH3CN | 26 |
| 3 | 2,4,6-Triphenylpyrylium Tetrafluoroborate | CH3CN | 18 |
| 4 | Eosin B | CH3CN | 21 |
| 5 | Eosin YS | CH3CN | 18 |
| 6 | Rhodamine B | CH3CN | 19 |
| 7 | p-toluenethiol | CH3CN | 46 |
| 8 | p-toluenethiol | H2O | 32 |
| 9 | p-toluenethiol | DMAc | 39 |
| 10 | p-toluenethiol | DMSO | 45 |
| 11 | p-toluenethiol | DCE | 53 |
| 12 | p-toluenethiol | THF | 43 |
| 13 | p-toluenethiol | 1,4-Dioxane | 51 |
| 14 | p-toluenethiol | IPA | 60 |
| 15 | p-toluenethiol | EtOH | 54 |
| 16 | p-toluenethiol | MeOH | 63 |
| 17c | p-toluenethiol | MeOH | 85 |
a Reaction conditions: 1,1-diphenylethylene (0.2 mmol, 35 μL), photocatalysts (20 mol%), solvents (1 mL), air, 30 W blue LED, r.t., 12 h. b Isolated yields. c O2. |
2.2 底物扩展
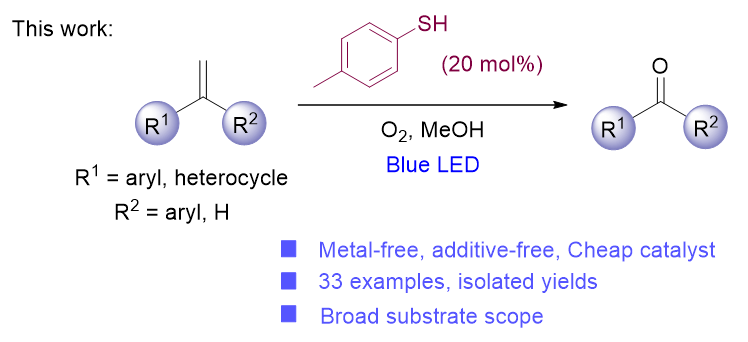
在确定烯烃氧化反应的最优条件后, 我们首先对单取代1,1-二芳基乙烯底物的普适性进行了考察(图2). 实验结果表明, 电子效应对该反应活性没有显著影响, 无论是带供电子基团(Me、Et、tBu、OMe、Ph)还是吸电子基团(F、Cl、CF3、NO2)的1,1-二芳基乙烯均能以中等至良好的收率(51%~90%)得到目标产物(2~10). 特别指出的是, 对位(2)、间位(11)和邻位(12)甲基取代底物的产率存在显著差异, 邻甲基取代的底物仅能以65%的收率得到目标产物, 可能是因为空间位阻的作用. 此外, 不同位置取代的含氟烯烃底物(7 vs. 13)均成功生成了相应的氧化产物, 为进一步的偶联衍生提供了可能性. 在标准反应条件下, 双取代和单取代的烯烃反应结果一致, 都具有良好的反应收率(14~16). 值得注意的是, 杂芳基烯烃的电子性质对反应的影响可忽略不计, 分别以43%和66%的收率获得相应的芳香酮(17, 18). 含烷基的烯烃底物反应也能顺利进行, 产率适中(19, 20). 随后, 我们在最优条件下分别以67%和69%的产率合成了药物分子酮洛芬甲酯(21)和非诺贝特(22). 非诺贝特[20]是一种良好的降血脂药物, 主要用于治疗异常的脂质水平.
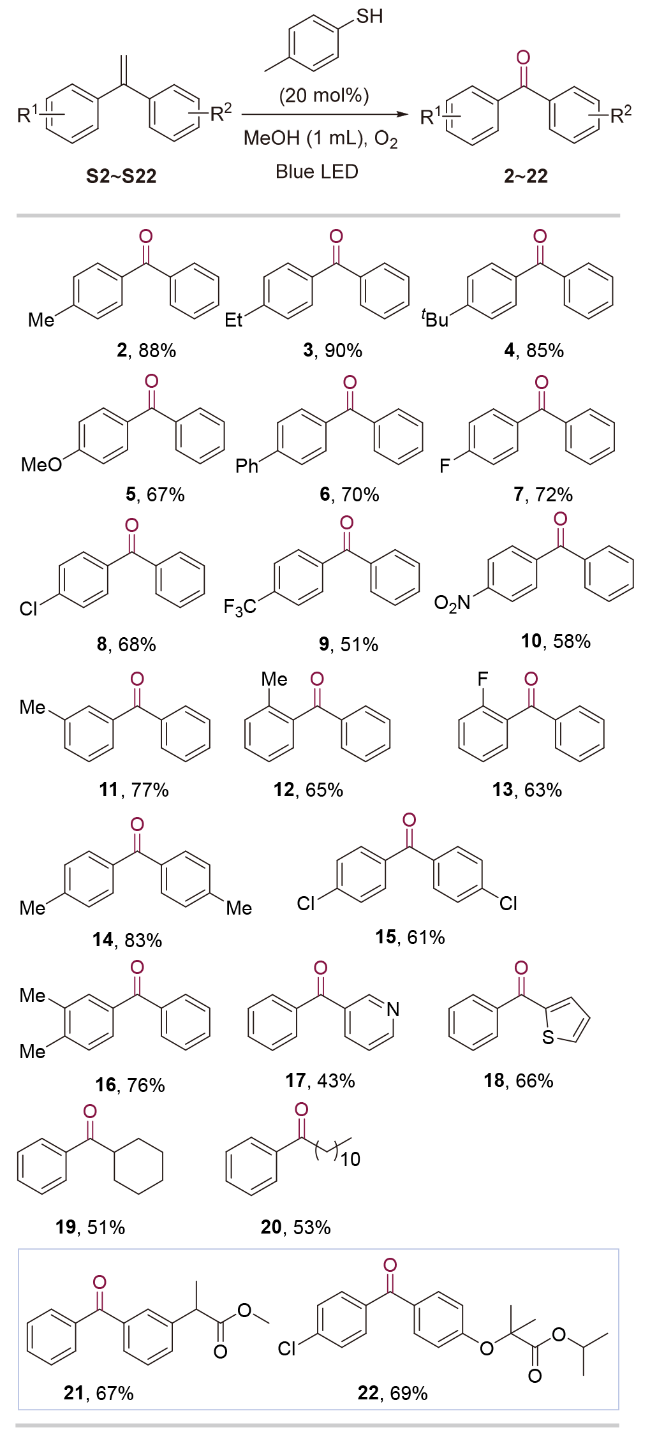
随后, 我们考察了不同α-烷基芳烯在最优条件下的适用性(图3). 具有中性(23)和富电子(24, 25)基团的α-烷基芳烯底物都能顺利反应, 生成相应的产物. 一系列吸电子(F、Cl、Br、I、CN和NO2)取代的α-烷基芳烯可以以中等至较高的产率生成相应的芳香酮(26~31). 此外, 稠环、吡啶、噻吩等杂环取代的α-烷基芳烯也适用于该氧化反应(32~34), 从而为合成杂环取代的芳香酮提供了有效方法(45%~65%). 以上结果表明, 该方法效率很高, 具有广泛的底物范围和良好的官能团容忍性.
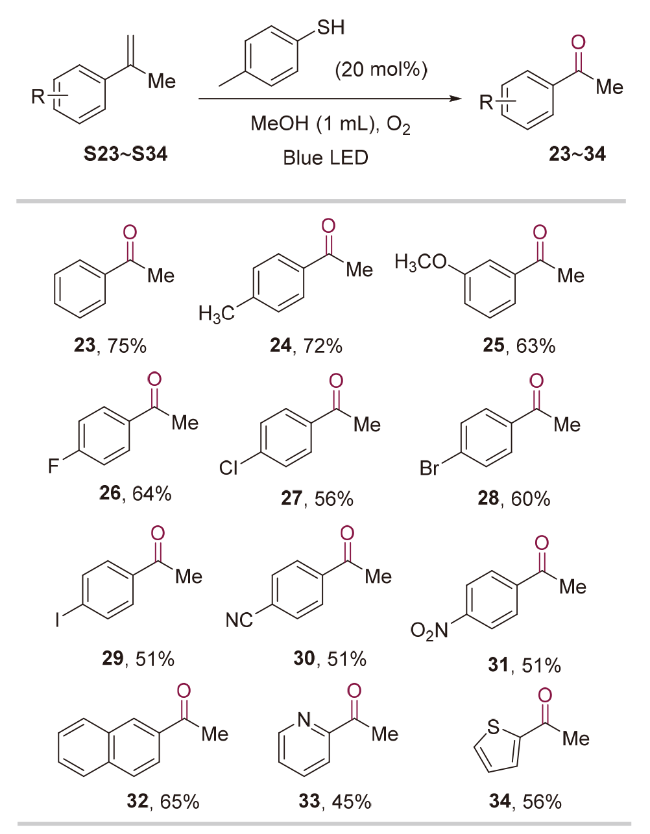
为了说明光催化烯烃C=C双键氧化裂解方法的潜在合成应用, 我们对2-苯基-1-丙烯的氧化反应进行了克级规模的放大量实验. 如图4所示, 在最优的反应条件下, 以2-苯基-1-丙烯S23 (8 mmol, 1.04 mL)为反应底物, 在甲醇溶液中以40倍的比例放大, 延长反应时间至48 h, 该反应非常顺利, 能以66%的产率获得产物苯乙酮23.
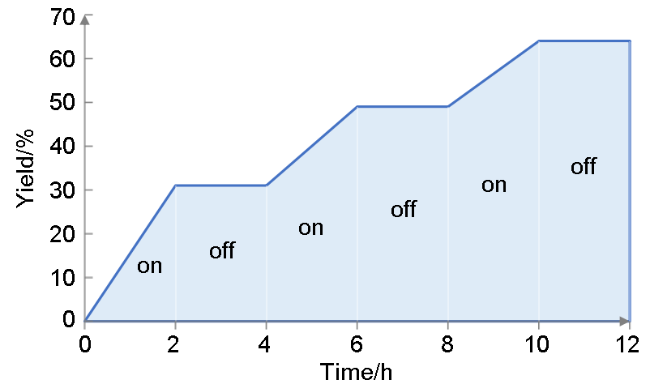
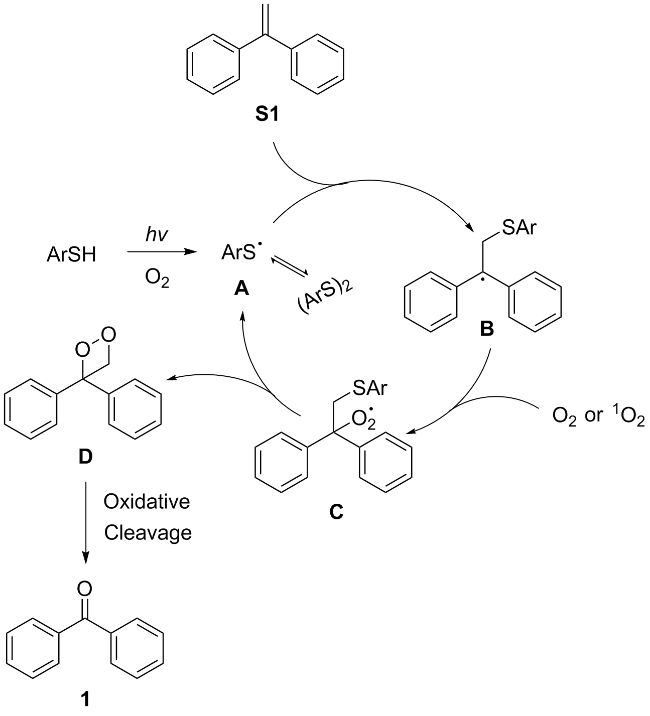
为了探究这种转变的可能机制, 我们进行了一系列的控制实验(图5). 在标准反应体系中, 加入自由基猝灭剂2,2,6,6-四甲基-1-哌啶二氧基(TEMPO)或2,6-二叔丁基-4-甲基苯酚(BHT), 完全阻止了该转化, 表明该反应可能经历自由基过程(图5, Eq. (a)). 当使用超氧自由基猝灭剂 2,2-二苯基-1-吡啶肼(DPPH)和苯醌时, 在体系中检测到目标产物二苯甲酮1, 表明了超氧自由基阴离子($O_{2}^{\centerdot -}$)未参与反应(图5, Eq. (b)). 随后, 加入单线态氧猝灭剂1,4-二氮杂环[2,2,2]辛烷(DABCO)或叠氮化钠(NaN3), 反应被完全抑制(图5, Eq. (c)), 表明单线态氧(1O2)可能参与了该氧化过程. 此外, 在无光照或氮气条件下, 该反应被完全抑制, 表明蓝光光照和氧气在烯烃的氧化过程中都必不可少(图5, Eqs. (d), (e)). 开关灯实验表明反应需要持续光照, 排除了链式反应机制的可能性(图6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
总之, 我们以4-甲苯硫酚作为光催化剂, 以分子氧作为绿色的氧化剂, 发展了一种高效、新颖的可见光催化烯烃C=C双键氧化裂解成酮的反应. 目前的催化体系具有反应条件温和、产率高、操作简单、官能团耐受性好且易于规模化等优点. 同时, 利用该方法合成了重要的药物分子酮洛芬甲基酯和非诺贝特, 突出了该方法的适用性和实用性.
4 实验部分
4.1 蓝光促进烯烃C=C双键氧化裂解反应的实验步骤
在空气中, 向一个干燥的25 mL的石英管中分别加入搅拌子, 烯烃(0.2 mmol), 4-甲苯硫酚(20 mol%, 5.0 mg)和甲醇(1 mL). 待试剂加入完毕后, 通入氧气, 在30 W蓝色LED灯照射下搅拌12~24 h(通过薄层色谱法检测). 将反应混合物冷却至室温, 用二氯甲烷洗涤溶液, 减压蒸馏除去溶剂后, 用制备薄层色谱板(石油醚/乙酸乙酯体积比20∶1)纯化残渣, 得到纯的产品.
(Cheng, B.)