

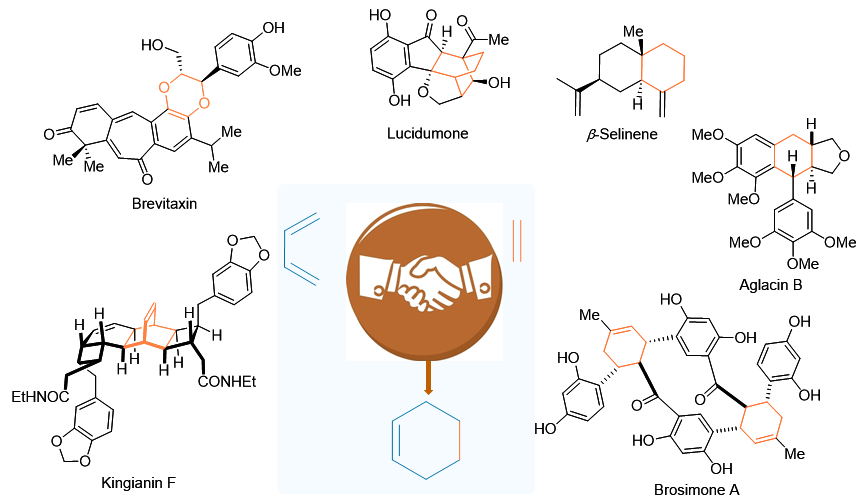
1 引言
复杂活性天然产物是新药研发的重要物质来源[1], 发展新颖、高效环加成反应, 实现复杂环系的立体选择性构建, 一直是合成化学家关注的研究热点之一[2]. 针对复杂环系的高效构建, 环加成反应的非对映选择性和区域选择性是重要的评价指标之一. 1928年, 德国化学家Otto Diels和Kurt Alder[3]首次报道了丁二烯(双烯体)与顺丁烯二酐(亲双烯体)经环加成反应得到环己烯产物, 该反应被命名为Diels-Alder反应(简称D-A反应). 根据Hückel分子轨道模型中双烯体和亲双烯体的相对能级差异(如HOMO: Highest Occupied Molecular Orbital, LUMO: Lowest Unoccupied Molecular Orbital), D-A反应分为正常电子需求D-A反应(即HOMOdiene- LUMOdienophile)和逆电子需求D-A反应(即LUMOdiene- HOMOdienophile)[4]. 根据Woodward-Hoffmann规则和前线轨道理论, 上述两种类型D-A反应均可在热条件下发 生[5]. 通常, 正常电子需求D-A反应的反应性由富电子双烯体和缺电子亲双烯体之间的HOMOdiene- LUMOdienophile相互作用调控. 相比之下, 逆电子需求D-A反应的反应性主要由HOMOdienophile-LUMOdiene相互作用主导[6]. 在D-A反应过程中, 经HOMO与LUMO相互作用, 由双烯体和亲双烯体的立体效应、电子效应、立体电子效应和次级轨道效应等, 调节D-A反应顺利发生, 并实现环化反应过程中反应性、立体选择性和区域选择性的可预测性[7]. 与此同时, 官能团化的双烯体和亲双烯体参与的D-A反应进一步拓展了其应用前景, 从而使得相应环加成反应得以顺利发生[8]. D-A反应以良好的立体选择性和区域选择性, 同时构建两根碳碳键和一个六元环, 进而推动D-A反应在活性复杂天然产物全合成中得到广泛应用(Scheme 1)[9], 奠定了其在周环反应发展过程中的历史地位[10].
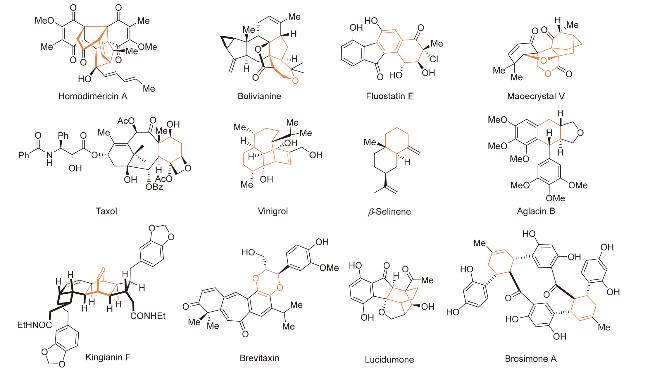
2 自由基介导Formal Diels-Alder反应
自由基介导亲双烯体和双烯体参与的formal Diels- Alder反应取得了一系列的发展. 1964年, Krauch课题 组[13]发现1,3-环己二烯在γ-辐射条件下经自由基途径发生[4+2]-环加成反应. 与此同时, 1968年, Ledwith课题组[14]报道N-vinylcarbazole在单电子氧化反应条件下, 经自由基正离子反应途径, 发生[2+2]-环加成反应, 从而使得自由基正离子介导的环加成反应进入研究视野. 之后, Bauld课题组[15]利用单电子氧化反应条件, 经自由基正离子中间体发生formal Diels-Alder-环加成反应, 并对环加成反应的底物适用性和可能的反应机理展开系统性研究. 自此, 自由基介导formal Diels-Alder反应受到有机合成化学家的广泛关注[16].
近年来, 基于自由基正离子中间体的光催化formal Diels-Alder环加成反应在方法学领域取得了显著进 展[20]. 研究历程如下所述: 2011年, Yoon课题组[21]首次报道了钌(II)联吡啶配合物在可见光照射下催化苯乙烯衍生物的单电子氧化过程, 生成的自由基正离子中间体可与富电子双烯体发生formal Diels-Alder环加成反应. 2015至2017年间, 多个研究团队(Ferreira[22]、Shores[23]、Brasholz[24]以及Nicewicz和Huang[25])相继发展了基于苯乙烯类底物的可见光催化自由基正离子环加成体系. 2017年, Zhao课题组[26]创新性地采用石墨相氮化碳
(g-C3N4)作为非金属光催化剂, 在氧气氛围及可见光条件下实现了该转化. 在催化体系拓展方面, 2018年, Honda课题组[27]开发了噻吨鎓盐有机光催化剂体系; Lei课题组[28]则设计了Fukuzumi吖啶盐/钴肟双催化系统, 成功实现了苯乙烯与炔烃的formal Diels-Alder环加成. 2019年, Okada课题组[29]报道了以TiO2为半导体光催化剂的紫外光驱动体系(λ=365 nm). 2020年, Zhang课题组[30]突破性地将聚甲基丙烯酸甲酯(PMMA)基交联聚合物材料作为可回收光催化剂, 为多功能催化材料设计提供了新思路. 近年来, 该领域研究持续深化: 2022年Hoshino课题组[31]采用噻吨烷催化剂实现了β-卤代苯乙烯的转化; Kang课题组[32]则发展了Fe(III)(btz)3配合物催化体系. 2023年, Pirovano课题组[33]使用2,4,6-三苯基吡喃鎓盐催化2-乙烯基吲哚的环加成; Ishihara课题 组[34]首次报道了铁(III)催化的不对称反应体系, 通过自由基正离子-手性抗衡阴离子对作用, 实现对映选择性控制. 最新进展包括: 2024年, Oh课题组[35]开发的二芳基二硒醚单电子转移催化体系; Wang课题组[36]开发的Ag3PO4半导体光催化系统; 以及2025年Kobayashi课题组[37]发展的三芳甲基阳离子催化策略. 在反应机理研究方面, Dang课题组[38]通过密度泛函理论(DFT)计算揭示了该转化的关键影响因素: 电子效应主导环加成反应的区域选择性, 而空间位阻与电子效应的协同作用决定了反应的化学选择性.
在方法学研究取得系统性进展的背景下, 自由基介导的formal Diels-Alder环加成反应以其温和的反应条件、良好的官能团容忍性和突出的立体选择性, 在活性复杂天然产物全合成领域受到广泛关注. 本文综述自由基介导的formal Diels-Alder环加成反应在复杂天然产物全合成中的应用进展, 基于反应过程中自由基中间体的引发机制差异, 将此类反应分为以下三种类型(Scheme 2): (1)自由基正离子介导的formal Diels-Alder反应; (2)分子间单电子转移引发的formal Diels-Alder环加成反应; (3)双自由基中间体参与的formal Diels-Alder环加成反应. 该分类为理解此类反应的机理及其在复杂分子构建中的应用提供理论框架.
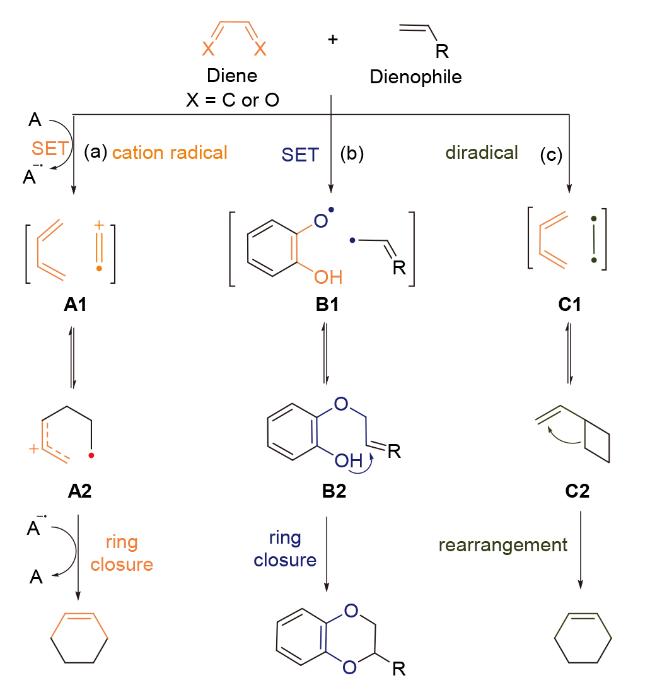
本综述将集中阐述自由基介导的formal Diels-Alder反应在天然产物全合成中的战略性应用, 重点讨论该反应的自由基引发机制、立体选择性控制要素以及当前存在的合成挑战.
3 天然产物的全合成应用
3.1 自由基正离子介导formal Diels-Alder反应的全合成研究
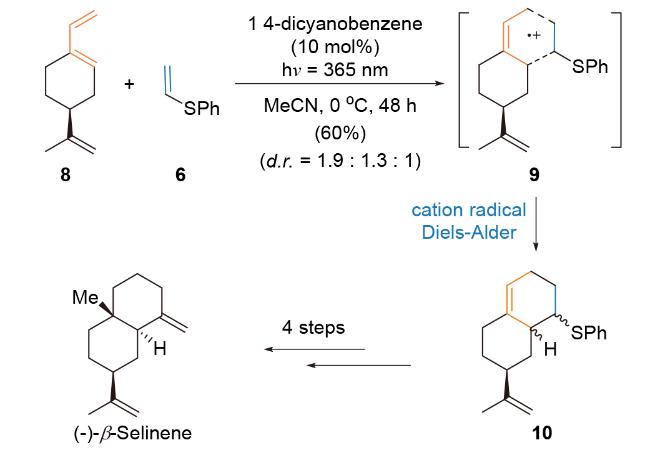
3.1.1 Bauld课题组(-)-β-Selinene的全合成研究
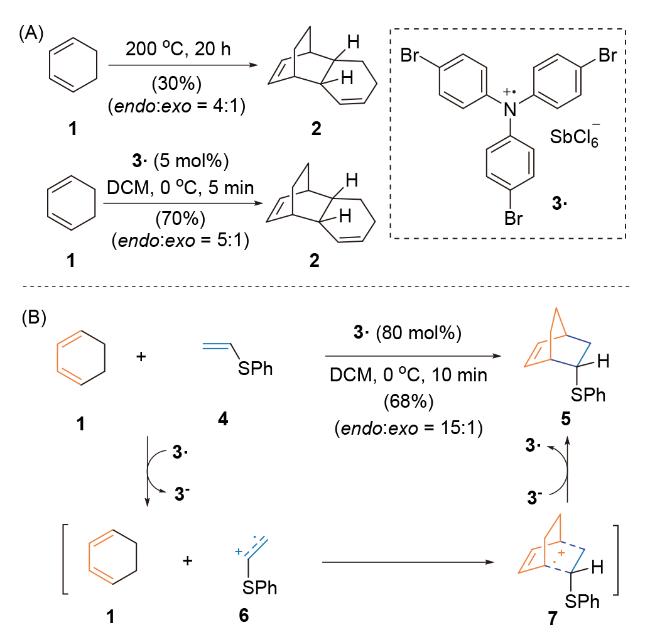
与此同时, Bauld课题组[40a]报道环己二烯1与富电子亲双烯体乙烯基硫醚4在自由基正离子盐氧化剂3●作用下, 经[4+2]-环加成反应, 生成环加成产物5. 作者猜测乙烯基硫醚(+1.42 V vs. SCE)比环己二烯1 (+1.53 V vs. SCE)更易被氧化, 生成自由基正离子中间体6, 进而与化合物1发生环加成反应, 生成自由基正离子中间体7, 该中间体进一步被还原, 得到稳定环加成产物5 (Scheme 3B). 经单电子氧化的自由基正离子活性中间体所参与formal Diels-Alder反应能够克服部分底物局限性, 实现非官能团化碳骨架的高效构建. 该方法提高了底物反应性、反应的非对映选择性和区域选择性, 仅需催化量的单电子氧化剂3●.
3.1.2 Sherburn课题组Kingianins的全合成
2013年, Sherburn课题组[44]利用自由基正离子介导formal Diels-Alder反应为关键合成策略, 完成了对天然产物Kingianins A, D和F的仿生全合成(Scheme 5). 炔类化合物11在Rieke-Zn的还原条件下, 化学选择性和非对映选择性得到(Z,Z,Z,Z)-四烯化合物11A[45], 进一步在100 ℃加热条件下触发串联8π-6π-电环化反应[46], 以1∶1的非对映选择性得到化合物12和13. 化合物13经Ley-Griffith氧化, 将羟基官能团转化为羧基官能团. 在催化量自由基正离子氧化剂3●作用下, 经过endo选择性的fomal Diels-Alder环加成反应, 生成化合物14和15. 通过接肽反应引入酰胺侧链得到天然产物Kingianins A (16)和D (17). 同时, 化合物12和13在催化量活化试剂3●作用下, 经endo选择性的fomal Diels- Alder反应, 以单一的非对映选择性得到环化产物19, 再经过羟基氧化为羧酸和接肽反应, 得到天然产物Kingianin F (20).
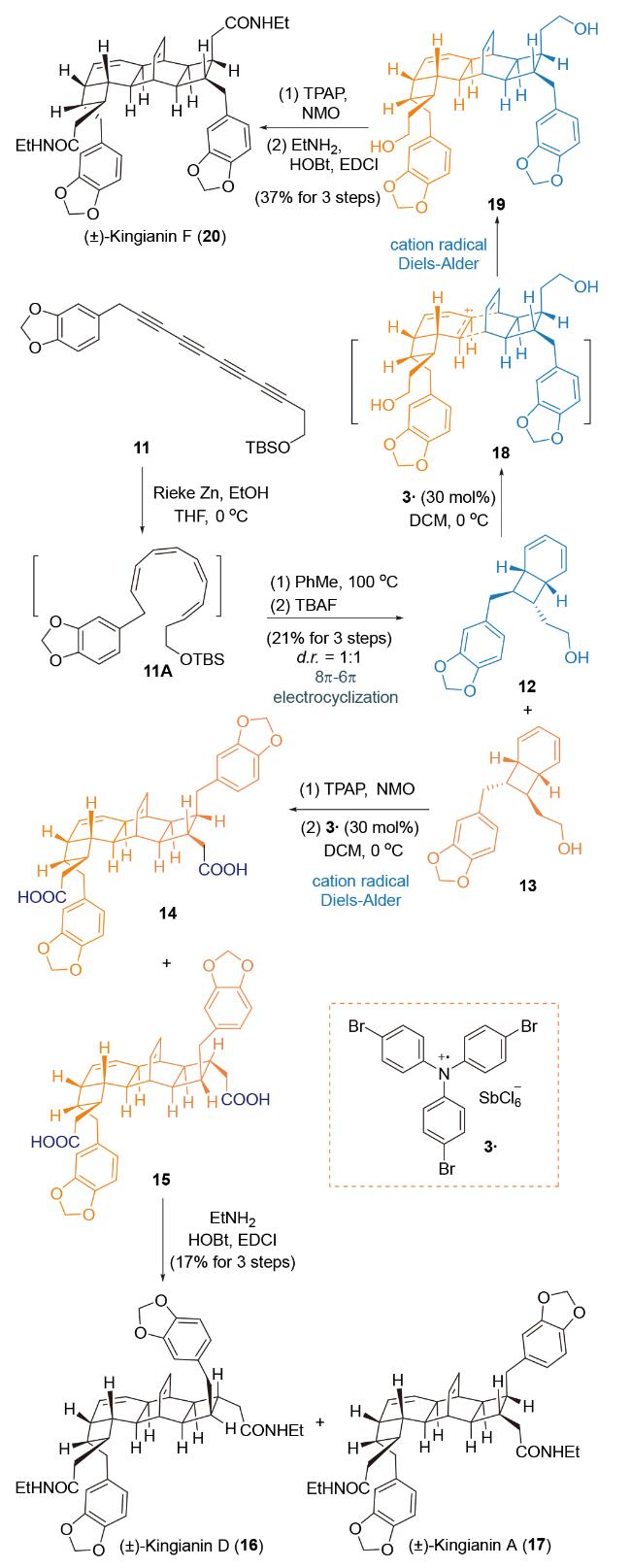
Sherburn课题组[47]将上述合成策略进一步应用于同源天然产物kingianic acid E、kingianins A, D和F的集群式全合成研究中.
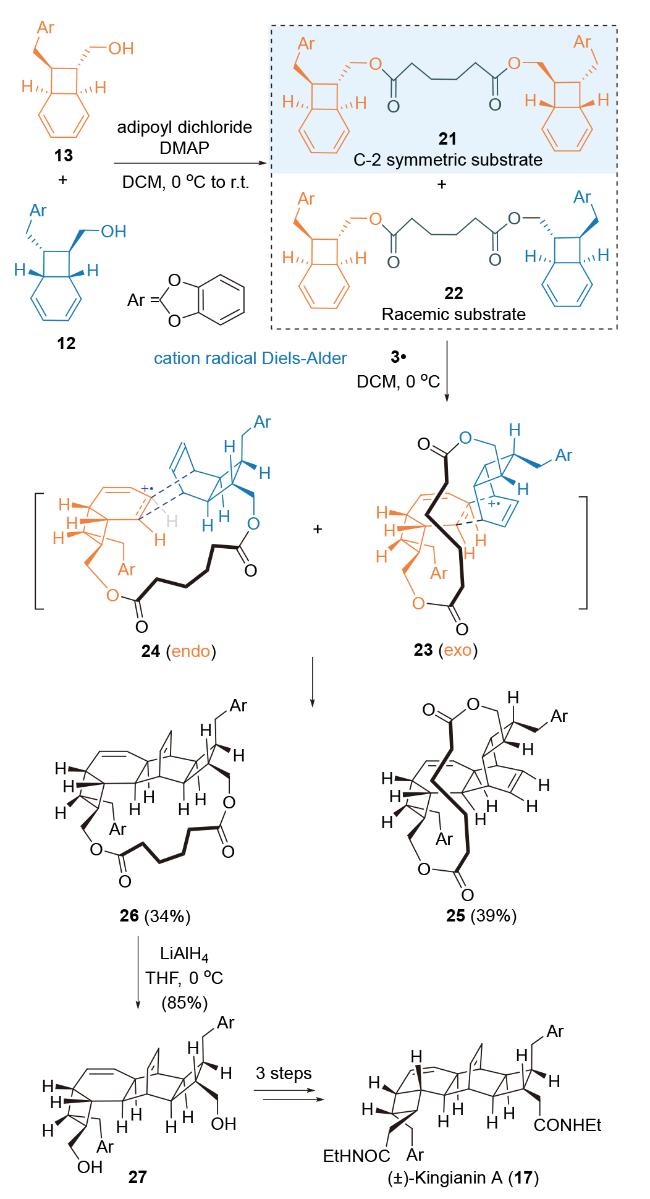
鉴于[4.2.0]辛二烯单体12和13在[4+2]-环加成反应过程中可能存在区域选择性和非对映选择性, Parker课题组[48]创造性使用己二酸-tethered合成策略, 利用分子内自由基正离子介导formal Diels-Alder反应为关键反应, 完成了对天然产物Kingianin A的非对映选择性全合成研究(Scheme 6). 在自由基正离子氧化剂3•的作用下, 链式前体化合物21未能发生[4+2]-环加成反应, 无法得到关环产物. 链式前体化合物22在该条件下发生分子内自由基正离子介导formal Diels-Alder反应, 分别得到环加成产物26 (34%)和25 (39%). 经环加成反应过渡态构象分析发现, exo-过渡态23中双烯体与亲双烯体之间的立体位阻较小, 反应更倾向得到环化产物25[49]. 环加成产物26再经四步转化, 得到目标天然产物(±)-Kingianin A.
3.1.3 Porco课题组Brosimones的全合成
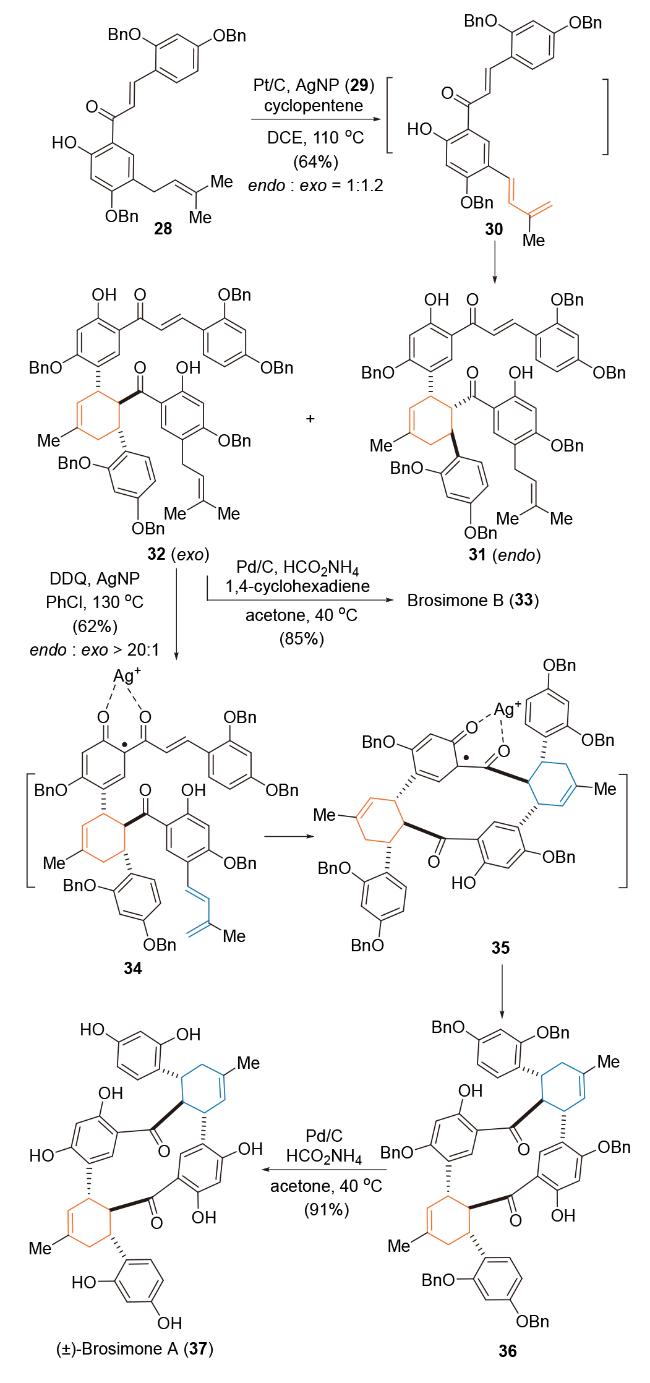
2013年, Porco课题组[53]利用串联脱氢-[4+2]-环加成反应为关键策略构建2'-氢查耳酮骨架, 完成了对天然产物Brosimones A和B的仿生全合成研究(Scheme 7). 化合物28在Pt/C-AgNP (AgNP=silver nanoparticles)协同催化条件下[54], 环戊烯作为储氢物种, 实现查耳酮骨架中异戊烯基的脱氢生成双烯体中间体30, 经自由基介导formal Diels-Alder反应, 以64%总产率和1∶1.2的endo/exo选择性, 得到环加成产物31和32. 化合物32脱除苄基保护, 得到Brosimone B. 与此同时, 化合物32在AgNPs/2,3-二氯-5,6-二氰基苯醌(DDQ)氧化条件 下[55], 得到自由基正离子中间体34, 经自由基介导formal Diels-Alder反应, 以62%的产率得到单一非对映异构体36, 进一步脱除苄基保护, 完成(±)-Brosimones A (37)的全合成研究. 该反应为该类家族天然产物提供了简洁的合成路线, 同时也证明在温和的反应条件下自由基介导串联脱氢-formal Diels-Alder反应在多环体系构建和复杂天然产物合成中的潜力[56].
3.1.4 祝介平课题组Aglacins的全合成
Podophyllotoxin和Picropodophyllotoxin是芳基四氢萘内酯木脂素类活性天然产物的代表, 其分别在并环处带有trans和cis的五元内酯环[57]. 与此同时, aglacins、gaultherin C、isoshonanin和sulabiroins A、B则是芳基四氢萘环醚木脂素类活性天然产物的代表, 其分别在并环处带有trans的五元醚环[58]. 该类活性家族天然产物展现出优良且多样的生物活性, 并已成功作为新药研发的先导化合物. 例如, 以podophyllotoxin为先导化合物进行的广泛构效关系研究开发出了抗癌药物——依托泊苷, 并被列入世界卫生组织(WHO)的基本药物清 单[59]. 因而, 开发简洁、可持续且成本效益高的合成方法, 用于制备带有稠合内酯或环醚的芳基四氢萘木脂素, 具有重要的药用价值.
2020年, 祝介平课题组[60]报道了以官能团化二肉桂醚38作为起始原料, 在Fukuzumi吖啶盐([Mes-Acr- Me]+BF4-)作为光催化剂, 通过蓝色LED照射, 以良好的产率和非对映选择性构建含有三个立体中心的[6,6,6,5]-芳基四氢萘环醚木脂素骨架结构(Scheme 8). 应用该类高效合成方法学, 从简单易得的单木脂素出发, 仅需两到三步转化, 成功实现六种天然产物: aglacin B、aglacin C、sulabiroin A、sulabiroin B、gaultherin C和isoshonanin的结构多样性合成, 并对gaultherin C的相对立体化学进行了修正和确认.
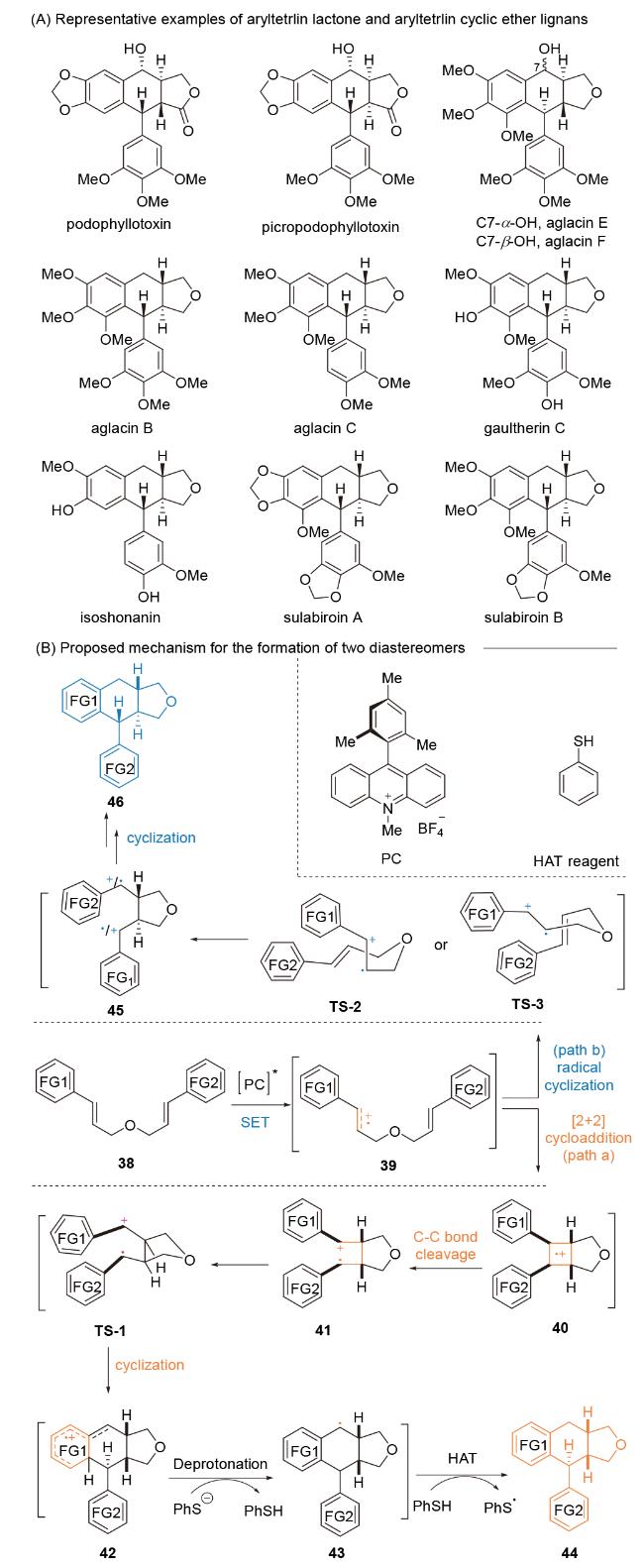
经反应机理研究, 作者对上述关键反应非对映选择性做出了系统深入阐述(Scheme 8B). 激发态[(Mes-Acr- Me)BF4]*对二肉桂醚39进行单电子氧化, 生成自由基正离子中间体39. 由反应路径A所示, 自由基正离子中间体39经分子内[2+2]-环加成反应[61], 生成cis-环丁烷自由基正离子中间体40, 之后经苄位C—C键(环丁烷中最弱的键)断裂, 由自由基正离子过渡态TS-1立体选择性芳基参与正离子环化反应或自由基环化反应, 生成自由基正离子中间体42. 42经苯硫酚负离子去质子化反应, 生成自由基中间体43和苯硫酚. 最后, 中间体43经分子间氢原子转移(HAT)反应会生成含cis-五元醚环的产物44.
由反应路径B所示, 自由基正离子中间体39经Beckwith-Houk模型, 通过过渡态TS-2或TS-3进行自由基环化反应, 生成trans-五元醚环自由基正离子中间体45[62]. 随后, 该中间体经芳基参与正离子环化反应或自由基环化反应, 生成含trans-五元醚环的产物46. 在此推测, 当底物38的芳香环上带有二甲氧基和三甲氧基时, 阳离子可能主要位于芳香环上, 从而降低了反应经自由基正离子参与的[2+2]-环加成反应可能性, 该反应由自由基参与的逐步环化反应为主, 最终生成含trans-五元醚环非对映异构体.
3.2 单电子转移参与的自由基介导formal Diels-Alder反应的全合成研究
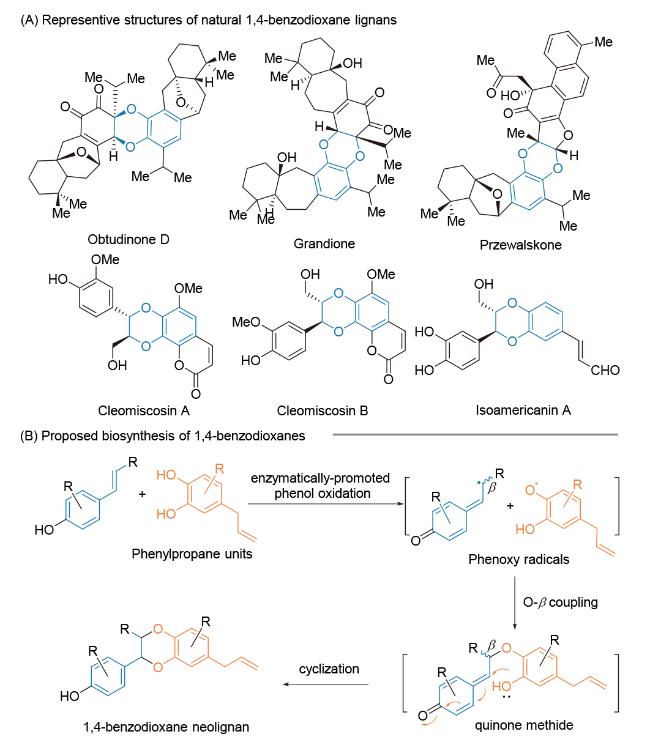
3.2.1 Ohira课题组的(±)-Aiphanol和(±)-nitidanin全合成研究
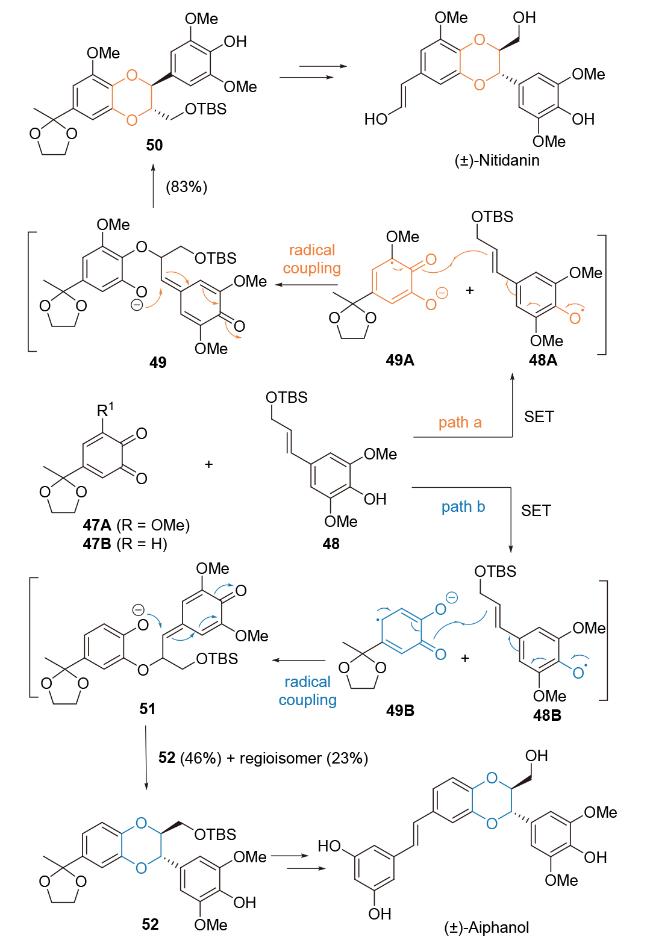
2003~2008年, Ohira课题组报道基于邻苯二醌参与的区域选择性分子间[4+2]-环加成反应对(±)- Aiphanol[70]和(±)-nitidanin[71]全合成研究. 在关键反应区域选择性机理研究中, 作者提出邻苯醌取代基效应对[4+2]-环加成反应区域选择性具有显著影响[72] (Scheme 10). 首先, 肉桂醇47的游离酚基和受保护的一级醇(TBS保护)是环加成反应成功进行的必要条件. 其次, 当在邻苯醌上添加甲氧基取代基时, 如化合物47A, 环加成反应的区域选择性显著提高(path a). 化合物47A和48经分子间单电子转移(SET), 生成自由基中间体48A和49A, 49A中甲氧基取代基能有效稳定相邻的碳自由基, 从而发生区域选择性自由基偶联反应, 生成中间体49, 进一步经共轭加成反应, 得到环加成产物50, 经后续官能团转化完成(±)-nitidanin全合成. 与此同时, 当邻苯醌47B参与反应时, 则以2∶1的区域选择性生成 [4+2]-环加成产物. 其中, 区域异构体52经官能团转化, 得到天然产物(±)-Aiphanol. 在该研究工作中, 邻苯二醌底物中取代基对基于电子转移的自由基介导fomal Diesl-Alder反应区域选择性影响, 为区域选择性构建木脂素类活性天然产物1,4-苯并二氧六环提供了新颖的合成策略.
3.2.2 黄俊课题组Brevitaxin的全合成
Brevitaxin最初由Arslanian等[73]于1995年从药用植物鼠尾草(Salvia)中以外消旋体形式分离出的icetexane-type二萜类活性天然产物(Scheme 11). 初步生物活性研究显示, 该化合物对MCF-7细胞(人乳腺癌细胞)具有显著的体外细胞毒性, 能选择性抑制前列腺癌细胞的增殖[74]. 在化学结构方面, Brevitaxin具有罕见的6/7/6/6四环骨架结构, 其高度不饱和的结构中包含一个trans-1,4-苯并二氧六环和一个环庚三烯酮结构单元. 生源合成假说提出, 该化合物的trans-1,4-苯并二氧六环可能由邻苯二醌前体54与松柏醇55参与的立体选择性hetero-Diels-Alder环加成反应构建[75].
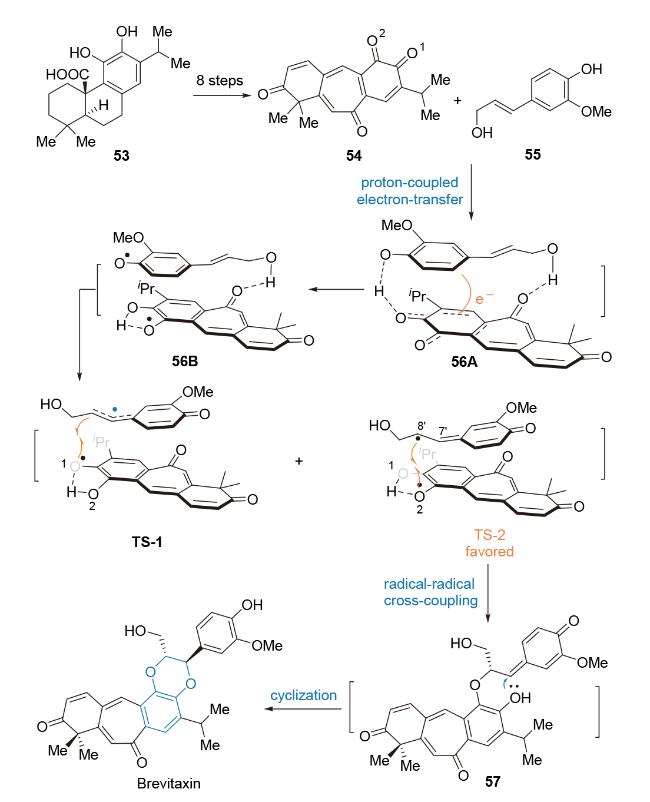
2024年, 黄俊课题组[76]以邻苯二醌参与的分子间 [4+2]-环加成反应为关键合成策略, 完成了对Brevitaxin的全合成研究(Scheme 11). 以化合物53为起始原料, 经八步转化得到化合物54, 该邻苯二醌化合物与化合物55经分子间[4+2]-环加成反应构建trans-1,4-苯并二氧六环结构单元. 对该反应机理研究显示: (1)化合物54和55之间通过分子间氢键相互作用, 经质子耦合电子转移(PCET)途径[77]形成双自由基中间体56B的反应能垒远低于经协同Diels-Alder环加成反应; (2)自由基偶联的区域选择性由酚氧基自由基的热力学稳定性所决定, 由于[6,7,6]-三环体系的共轭骨架结构使得O2自由基中间体较O1自由基中间体更稳定, 因此自由基偶联反应经过渡态TS-2进行, 生成中间体57经共轭加成反应, 得到环加成产物Brevitaxin.
在活性天然产物Brevitaxin的全合成研究中, 邻苯二醌参与的formal Diels-Alder反应中PCET机理的提出和验证为后续对于hetero-Diels-Alder反应的机理研究提供了新思考.
3.3 双自由基参与的formal Diels-Alder反应的全合成研究
3.3.1 胡安华课题组Lucidumone的全合成
Lucidumone于2019年从传统中草药——灵芝(Ganoderma lucidum)中分离出的萜类化合物[78]. 化学结构上, 该天然产物具有独特的6/5/6/6/5笼状五环结构, 包含双环[2.2.2]-结构单元上分布六个连续的立体中心, 其高氧化态包含一个对苯酚、一个二级羟基和两个酮羰基. 初步生物活性研究表明, Lucidumone能通过与Tyr385和Ser530残基直接结合, 选择性抑制COX-2, 具有潜在治疗炎症的药用价值. 值得注意的是, 在前期全合成研究中, 分子内Diels-Alder环加成反应已被成功应用于构建含有多个连续手性中心的双环[2.2.2]-结构单元[79].
可见光诱导环加成一直是绿色化学合成的标杆之一. 近年来被陆续报道通过单电子氧化剂、或者可见光催化剂氧化途径形成自由基正离子中间体, 进而引发formal Diels-Alder环加成反应. 与之不同的是, 通过光诱导能量转移途径生成双自由基中间体, 进而引发[4+2]-环加成反应的例子鲜有报道.
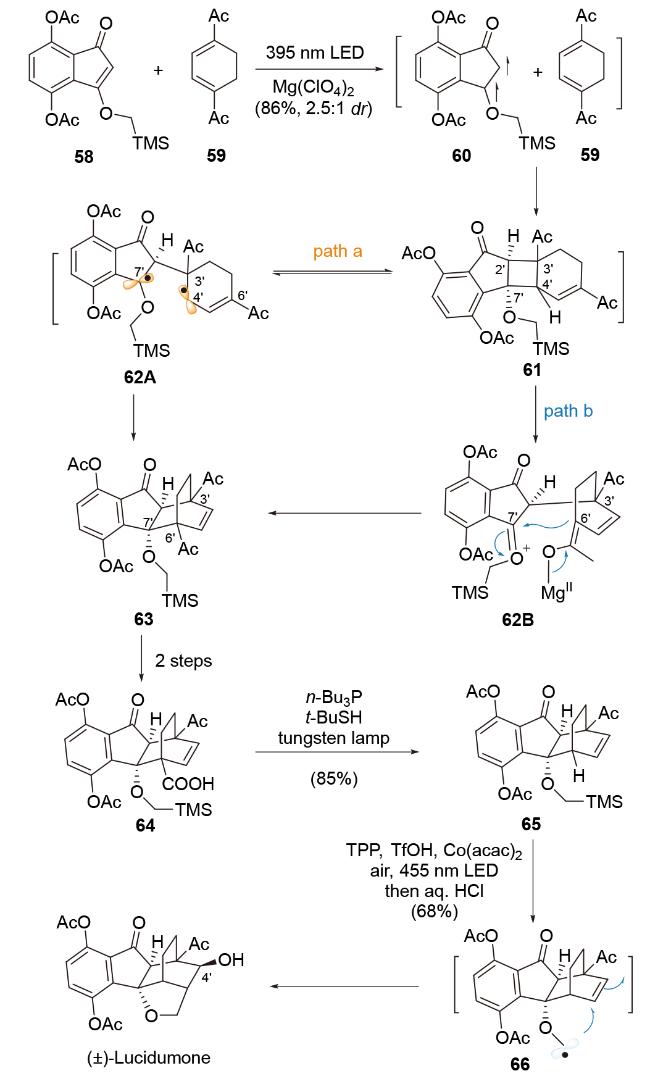
2024年, 胡安华课题组[80]利用光诱导的[4+2]-环加成反应为关键合成策略, 构建双环[2.2.2]-结构单元, 完成天然产物Lucidumone的全合成(Scheme 12). 在该全合成研究中, 烯烃底物58在光诱导反应条件下形成双自由基中间体60, 该中间体与双烯体59发生分子间非对映选择性[2+2]-环加成反应, 构建环丁烷结构单元61, 该环丁烷中间体在路易斯酸介导的光照反应条件下, 可能经历两种重排反应途径构建双环[2.2.2]-结构单元. 首先, 原位生成的双自由基物种62A可由中间体61发生C2'—C4'键断裂, 触发目标重排反应路径a, 生成C4'—C7'键, 得到化合物63. 其次, 经重排反应路径b, 在外加路易斯酸存在下成功生成两性离子中间体62B, 进一步经环化反应, 得到化合物63. 之后, 经三步官能团转化, 脱除C6'的乙酰基官能团, 得到化合物65. 最后, 通过光催化氧化烷氧甲基化反应[81], 实现多环体系中五元醚环和C4'-OH的引入, 最终完成Lucidumone的全合成研究.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
该[4+2]-环加成反应通过新颖的光诱导能量转移途径, 引发串联的分子间[2+2]-环加成反应和重排反应, 成功拓展缺电子双烯体参与自由基介导formal Diels-Alder反应的反应模式.
4 小结与展望
在温和的反应条件下进行的自由基介导formal Diels-Alder反应, 凭借其优异的区域选择性、非对映选择性和对映选择性、良好的官能团兼容性, 在复杂活性天然产物全合成研究中展现独特的高效性. 与此同时, 丰富的理论计算和机理验证实验对于自由基介导formal Diels-Alder反应过程中的可能反应机理提供了夯实的理论基础和依据. 在此, 作者提出如下思考.
大自然作为鬼斧神工的造物主, Diels-Alder反应涉及众多活性天然产物生源合成途径之中. 近年来, 随着合成生物学的蓬勃发展, 一系列Diels-Alder酶被分离报道[82], 反应机理的深入研究表明, 协同和分步的D-A反应机理均有可能存在. 因此, 新的D-A酶的发现, 以及在酶催化反应条件下, formal Diels-Alder环加成反应机制探索具有重要科学意义.
近年来, 可见光催化及有机电化学等新型反应模式的快速发展, 提供了更加丰富的自由基活性中间体引发方式, 为自由基介导formal Diels-Alder反应提供潜在新颖的合成途径.
(Cheng, F.)