

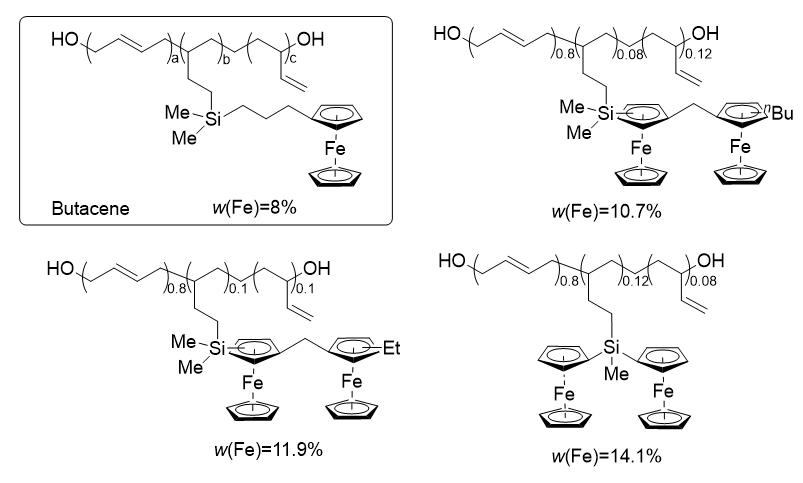
1 引言
2 结果与讨论
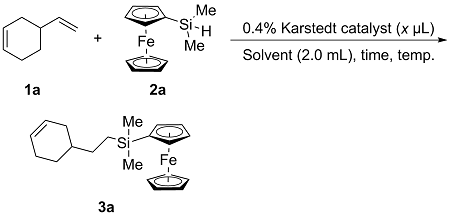
2.1 含硅双核二茂铁衍生物与烯烃的加成反应优化
表1 烯烃与二茂铁硅烷加成模板反应条件优化Table 1 Optimization of reaction conditions for ferrocene silane and olefin |
| Entrya | 催化剂用量 | 溶剂 | 反应时间/h | 反应温度/℃ | 收率/% |
|---|---|---|---|---|---|
| 1 | 5 μL | 甲苯 | 16 | 60 | 22.9 |
| 2 | 50 μL | 甲苯 | 16 | 60 | 75.5 |
| 3 | 100 μL | 甲苯 | 16 | 60 | 82.4 |
| 4 | 200 μL | 甲苯 | 16 | 60 | 73.4 |
| 5 | 100 μL | THF | 16 | 60 | 78.1 |
| 6 | 100 μL | DCM | 16 | 60 | 70.6 |
| 7 | 100 μL | 甲苯 | 4 | 60 | 81.5 |
| 8 | 100 μL | 甲苯 | 8 | 60 | 86.3 |
| 9 | 100 μL | 甲苯 | 24 | 60 | 75.8 |
| 10 | 100 μL | 甲苯 | 8 | 40 | 82.9 |
| 11 | 100 μL | 甲苯 | 8 | 80 | 78.8 |
| 12 | 100 μL | 甲苯 | 8 | 100 | 77.4 |
a Reaction conditions: 1a (0.82 mmol, 2.0 equiv.), 2a (0.41 mmol, 1.0 equiv.), 0.4% Karstedt catalyst, solvent (2.0 mL), temperature, time; Yield was calculated based on dimethyldiferrocenylsilane. |
2.2 HTPB接枝含硅二茂铁衍生物的合成
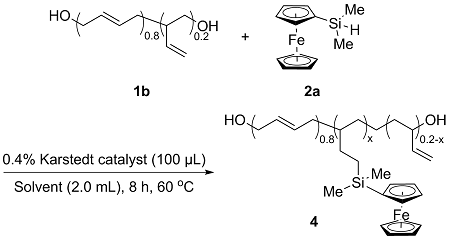
2.2.1 二甲基二茂铁硅烷与HTPB的模板反应研究
表2 双核二茂铁硅烷与HTPB的硅氢化反应Table 2 Hydrosilylation reaction of biferrocene silane and HTPB |
| Entrya | 产物编号 | 二茂铁硅烷与HTPB物质的量比 | 收率/% | 接枝率b/% | 铁含量c/% |
|---|---|---|---|---|---|
| 1 | 4a | 5∶1 | 83 | 13.6 | 4.2 |
| 2 | 4b | 10∶1 | 97 | 29.2 | 8.3 |
| 3 | 4c | 15∶1 | 94 | 41.6 | 9.0 |
| 4 | 4d | 20∶1 | >99 | 58.3 | 10.2 |
| 5 | 4e | 25∶1 | >99 | 66.7 | 11.1 |
| 6 | 4f | 30∶1 | >99 | 79.2 | 12.9 |
a Reaction conditions: HTPB (0.02 mmol, 2.0 equiv.), 2a, 0.4% Karstedt catalyst (100 μL), toluene (2.0 mL), 60 ℃, 8 h; Yield calculated based on dimethyldiferrocenylsilane; b The grafting ratio was calculated from the 1H NMR; c the iron content was calculated inversely from elemental analysis. |
2.2.2 HTPB接枝含硅二茂铁衍生物的合成
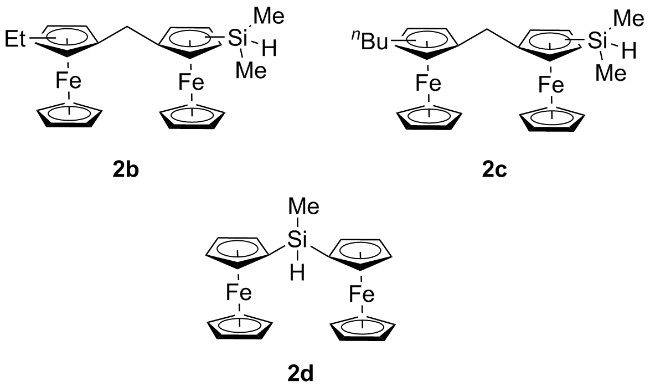
表3 双核二茂铁硅烷2b与HTPB的硅氢化反应Table 3 Hydrosilylation reaction of biferrocene silane 2b and HTPB |
| Entrya | 产物编号 | 二茂铁硅烷与HTPB物质的量比 | 收率/% | 接枝率b/% | 铁含量c/% |
|---|---|---|---|---|---|
| 1 | 5a | 5∶1 | 93 | 12.5 | 3.1 |
| 2 | 5b | 10∶1 | 90 | 25.0 | 7.0 |
| 3 | 5c | 15∶1 | 88 | 33.3 | 8.7 |
| 4 | 5d | 20∶1 | 79 | 50.0 | 11.2 |
| 5 | 5e | 30∶1 | 61 | 50.0 | 11.9 |
| 6 | 5f | 40∶1 | 48 | 45.8 | 10.4 |
a Reaction conditions: HTPB (0.02 mmol, 2.0 equiv.), 2b, 0.4% Karstedt catalyst (100 μL), toluene (2.0 mL), 60 ℃, 8 h; Yield calculated based on dimethyldiferrocenylsilane; b The grafting ratio was calculated from the 1H NMR; c The iron content was calculated inversely from elemental analysis. |
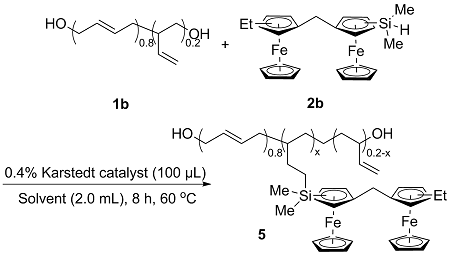
表4 双核二茂铁硅烷2c与HTPB的硅氢化反应Table 4 Hydrosilylation reaction of biferrocene silane 2c and HTPB |
| Entrya | 产物编号 | 二茂铁硅烷与HTPB物质的量比 | 收率/% | 接枝率b/% | 铁含量c/% |
|---|---|---|---|---|---|
| 1 | 6a | 10∶1 | 79 | 25.0 | 8.8 |
| 2 | 6b | 20∶1 | 55 | 33.3 | 9.4 |
| 3 | 6c | 30∶1 | 52 | 45.8 | 10.7 |
| 4 | 6d | 40∶1 | 51 | 41.7 | 10.6 |
a Reaction conditions: HTPB (0.02 mmol, 2.0 equiv.), 2c, 0.4% Karstedt catalyst (100 μL), toluene (2.0 mL), 60 ℃, 8 h; Yield calculated based on dimethyldiferrocenylsilane; b The grafting ratio was calculated from the 1H NMR; c The iron content was calculated inversely from elemental analysis. |
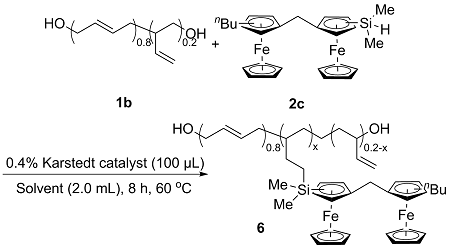
表5 双核二茂铁硅烷2d与HTPB的硅氢化反应Table 5 Hydrosilation reaction of biferrocene silane 2d and HTPB |
| Entrya | 产物编号 | 二茂铁硅烷与HTPB物质的量比 | 收率/% | 接枝率b/% | 铁含量c/% |
|---|---|---|---|---|---|
| 1 | 7a | 10∶1 | 97 | 20.8 | 8.3 |
| 2 | 7b | 20∶1 | 80 | 33.3 | 11.2 |
| 3 | 7c | 30∶1 | 75 | 45.8 | 11.9 |
| 4 | 7d | 40∶1 | 72 | 58.3 | 14.1 |
a Reaction conditions: HTPB (0.02 mmol, 2.0 equiv.), 2d, 0.4% Karstedt catalyst (300 μL), toluene (2.0 mL), 60 ℃, 8 h; Yield calculated based on dimethyldiferrocenylsilane; b The grafting ratio was calculated from the 1H NMR; c The iron content was calculated inversely from elemental analysis. |
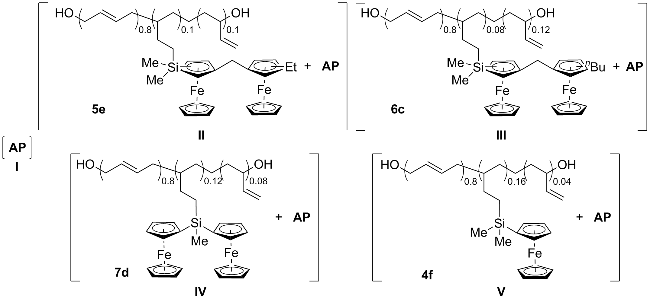
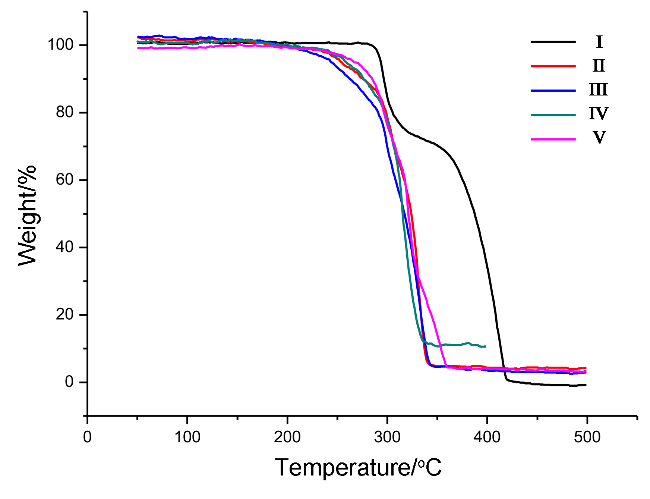
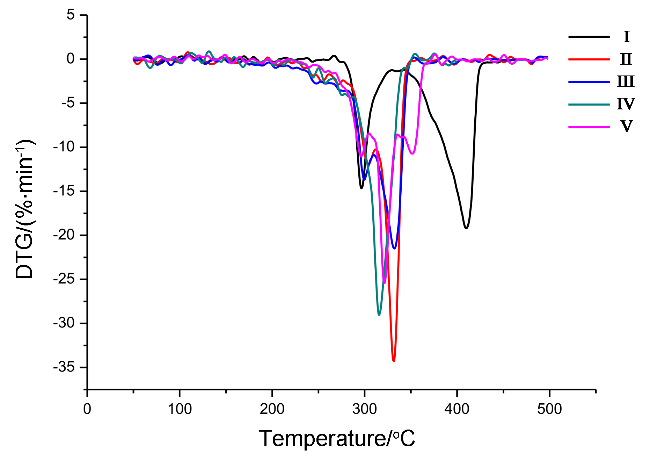
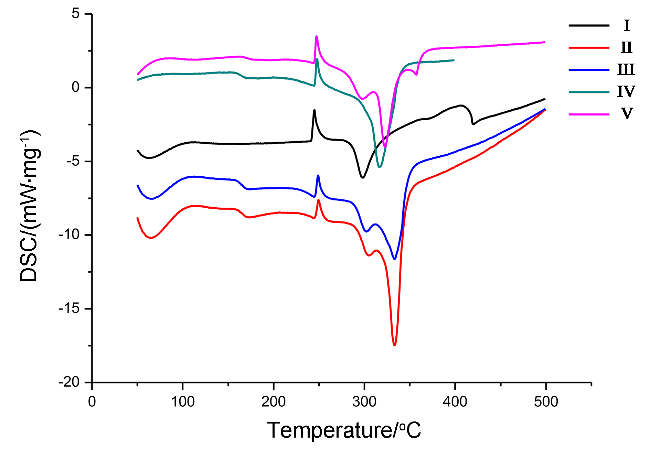
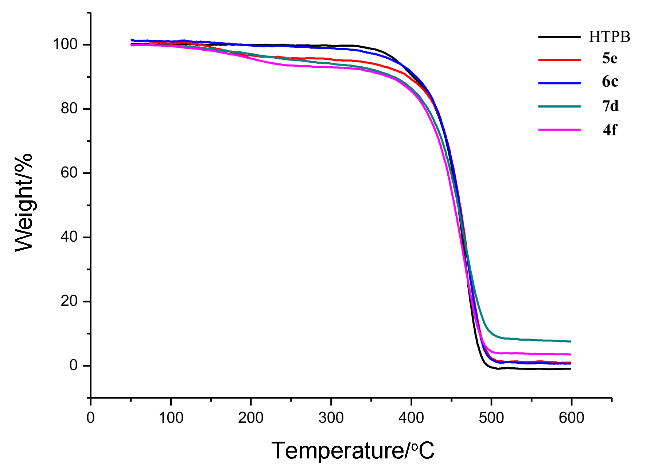
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}