

1 引言
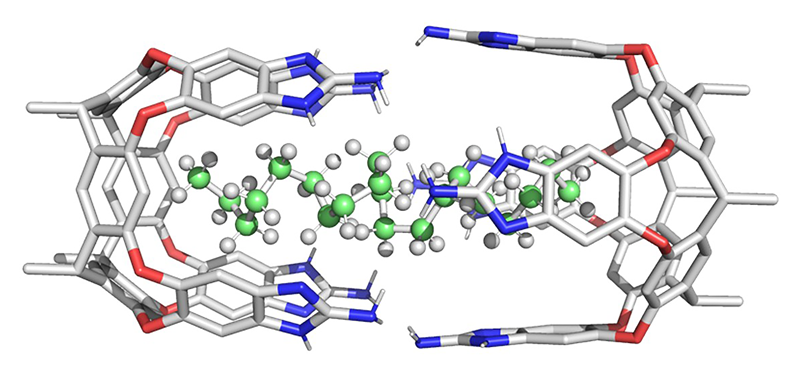
非共价分子胶囊因具备与酶活性中心相似的疏水微环境[8], 在底物分子预组织方面展现出独特的优势, 成为超分子化学研究的热点. 这类分子胶囊通常以杯[4]芳烃和间苯二酚杯[4]芳烃等凹面分子为骨架, 通过氢键、卤键、硫族键、疏水或静电等非共价相互作用, 自组装形成封闭结构[9-10], 进而调控其空腔内部分子的构象. 2003年, Reinhoudt等[11]首次报道了pH=9 缓冲溶液中, 上缘分别带有羧酸根阴离子与胍基阳离子的杯[4]芳烃, 通过静电作用形成杂二聚体分子胶囊, 实现对N-甲基奎宁环鎓氯化物的特异性包结. 2004年, Gibb 等[12]基于形状互补与疏水作用协同, 构建由深穴刚性间苯二酚杯[4]芳烃组成的分子胶囊, 能够高效包裹甾类分子或与直链烷烃形成2∶1、2∶2的主客体复合物. 2013年, Rebek等[13]则通过在苯并咪唑酮基间苯二酚杯[4]芳烃下缘引入水溶性吡啶盐基团, 借助上缘氢键作用, 在合适客体如柔性的直链烷烃或刚性的二苯乙烯类客体分子存在下形成氢键胶囊.
在上述非共价分子胶囊的传统研究范式中, 分子胶囊被视作“静态容器”, 凭借其预先构筑的限域空间对客体分子的构象进行调控. 通过氢键、 疏水作用等非共价相互作用, 这类分子胶囊能够实现对客体分子的选择性识别与功能转化[14-18]. 尽管该策略在分子识别、催化等领域取得了显著进展, 但其核心逻辑仍依赖主体分子的预设结构, 而对客体分子自身构象变化在驱动分子胶囊形成中的作用关注不足. 这种单向的调控模式, 在一定程度上限制了超分子组装体系的动态响应性与设计灵活性. 值得注意的是, 直链烷烃分子在不同环境中的构象行为呈现显著差异. 在溶液中, 直链烷烃通常以能量较低的线性伸展构象存在, 然而, 当进入分子胶囊的限域空间后, 受疏水效应与C—H…π等非共价相互作用的协同影响, 其碳-碳键会发生邻位交叉构象转变, 进而呈现弯曲、螺旋等特殊形态[19-20]. 这种构象变化在生物体系中广泛存在, 例如细胞膜磷脂双分子层中的脂肪酸链[21-22], 通过动态构象调整维持膜结构的稳定性与流动性. 但在人工合成的非共价分子胶囊体系中, 客体分子的这种构象适应性变化尚未被充分挖掘与利用, 如何将客体构象变化与主体自组装过程相耦合, 是本工作重点关注的问题. 基于此, 本工作以间苯二酚杯[4]芳烃为骨架, 通过加深芳香壁、修饰上缘功能基及引入下缘水溶性基团, 合成了2-氨基苯并咪唑基分子杯1. 研究发现, 当对称型直链烷烃(n-C12H26~n-C20H42)及非对称型苯乙烯类半菁染料分子SP-Cn (n=1~12)作为客体时, 其在疏水空腔内可自发调整为伸展、盘绕或弯曲折叠等多样化构象, 并通过构象自适应与主体分子产生协同疏水作用及C—H…π作用, 驱动分子杯1发生二聚, 形成稳定的非共价分子胶囊. 这一发现揭示了客体构象变化对主体自组装的关键诱导作用, 为非共价分子胶囊的设计与构建提供了全新策略.
2 结果与讨论
2.1 合成与结构
2-氨基苯并咪唑基分子杯1的合成参考已报道的方法[23], 其具体合成路线如Scheme 1(a)所示. 以酯基修饰的八氨基分子杯2为起始原料, 通过与溴氰发生反应, 随后进行碱性条件下的酯基水解, 得到目标产物1. 分子杯1在水相中呈现出两种典型构象, 可通过核磁共振氢谱(1H NMR)进行区分(见支持信息图S2). 当间苯二酚杯[4]芳烃母体中的次甲基质子信号出现在δ 4.0时, 分子杯的四个芳香壁呈展开状态, 并以二聚体的形式存在, 称之为风筝构象; 而当次甲基质子信号位移至δ 5.5 时, 四个芳香壁垂直于底部的间苯二酚杯[4]芳烃母体, 称之为花瓶构象. 这两种构象的动态平衡受溶液环境、温度及客体分子的影响, 为后续主客体识别研究提供了结构基础[24]. 分子杯1的化学结构通过1H NMR、核磁共振碳谱(13C NMR)及高分辨质谱(HRMS)等分析手段进行验证. 半菁类染料分子SP-Cn的合成路线如Scheme 1(b)所示, 通过取代反应与Aldol缩合反应分步构建, 具体合成步骤及表征数据将在支持信息部分详细阐述.
2.2 对称型直链烷烃
核磁共振技术作为探究主客体相互作用的核心手段, 在本工作中发挥了关键作用. 向分子杯1的重水溶液中加入0.5 equiv.的直链烷烃, 经超声处理后进行1H NMR测试(如图1b). 研究发现, 短链的正庚烷和正十烷与分子杯1形成1∶1的自组装结构(见支持信息图S5). 对于正十一烷, 其主客体体系的核磁氢谱在高场区未出现封装信号峰, 这归因于正十一烷的体积与分子杯1、胶囊1.1的尺寸存在显著差异——其体积远小于胶囊 1.1, 又明显大于分子杯1的单腔容纳能力, 致使正十一烷无法被有效包结(见支持信息图S5). 当客体为正十二烷时, 其在高场区呈现6组宽化的包结信号峰, 表明正十二烷在胶囊1.1中采取对称伸展构象(如图1a所示),并通过快速运动适配疏水空腔[25]. 随着烷烃碳链增长, 高场区主客体核磁信号峰逐渐尖锐化, 这一现象揭示了主客体复合物动力学稳定性的提升[26].
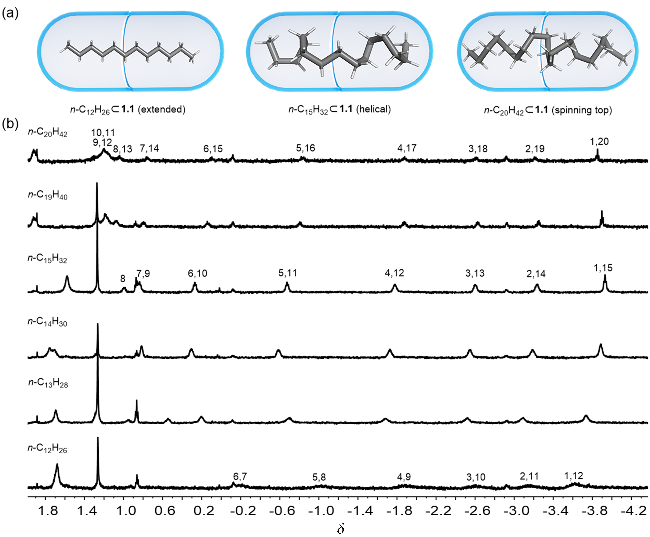
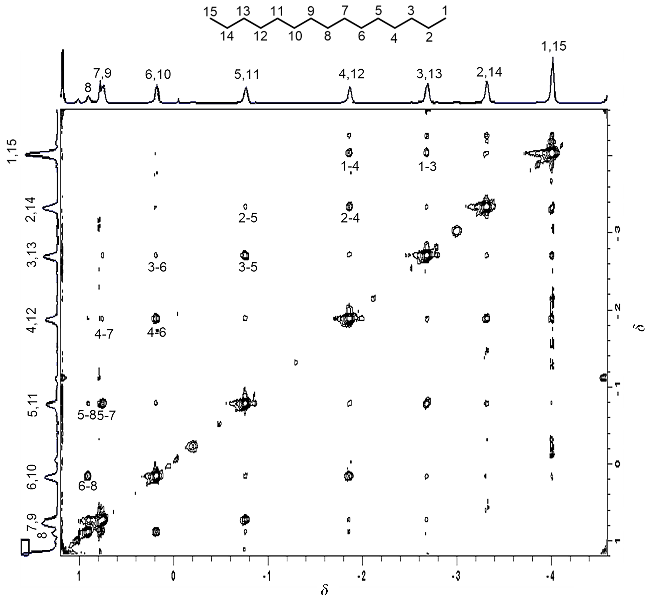
以正十五烷为例, 其较正十二烷能更好适配胶囊 1.1的疏水空腔, 末端甲基深入空腔最深处, 受芳香壁磁屏蔽效应影响, 化学位移变化值(Δδ)达-4.80. 正十五烷与胶囊1.1的二维核欧沃豪斯效应谱(2D NOESY)实验结果如图2所示, 被封装的正十五烷的C(1)与C(3)和C(4), C(2)与C(4)和C(5), C(3)与C(5)和C(6), C(4)与C(6)和C(7), C(5)与C(7)和C(8)有明显的交叉信号峰, 但是C(6)仅与C(8)有交叉信号峰, 没有观察到其与C(9)的NOE效应. 这表明正十五烷在空腔中呈螺旋—延展—螺旋的构象(如图1a所示). 扩散排序谱(DOSY)常用来估算分子的流体力学体积[27]. 为了进一步探究正十五烷与胶囊1.1的自组装状态, 我们向1中加入等物质的量的正庚烷和正十五烷, 然后进行DOSY测试(见支持信息图S7), 测得正庚烷与1的主客体复合物扩散系数D=2.02×10−6 cm2•s−1, 测得正十五烷与1的扩散系数D=1.54×10−6 cm2•s−1, 根据Stokes-Einstein方程可估算出, 正十五烷与1的流体力学体积是正庚烷与1的2.26倍, 这也进一步证实了正十五烷与1是1∶2的胶囊结构.
从构象演变的整体趋势分析, 与短链烷烃相比, 长链烷烃的化学位移呈现持续向高场偏移的特征. 这一现象表明, 长链烷烃在胶囊1.1的疏水限域空间内, 通过从分子两端向中间压缩的构象调整策略以适配空腔尺寸, 进一步证实了客体分子构象自适应对驱动分子杯二聚形成胶囊的关键作用.
2.3 非对称型直链烷基苯乙烯类半菁染料分子SP-Cn
相较于前述对称型直链烷烃分子, 烷基修饰的苯乙烯类半菁染料分子的非对称结构引入了刚性芳环与可调控的柔性烷基链双重特征, 为探究构象适应性驱动的超分子组装机制提供了更丰富的研究维度. 苯乙烯类半菁荧光染料分子(SP-Cn)因其独特的光学性质和与生物大分子的高亲和力而备受关注, 该类分子可通过静电作用与π-π堆积效应嵌入DNA碱基对中[30], 常作为荧光探针应用于DNA检测领域. 在本工作中, 我们设计合成了一系列具有不同碳链长度的非对称型直链烷基苯乙烯类半菁荧光染料分子(SP-Cn), 系统考察其与胶囊1.1的自组装行为, 旨在探究烷烃链段在胶囊疏水空腔内的构象演变规律, 以及主客体相互作用对SP-C1光物理性质的影响.
向SP-C1的重水溶液中加入1.0 equiv.的分子杯1时, 核磁氢谱(见支持信息图S10)显示两组主客体复合物特征信号峰: 一组峰为1∶1包结复合物(SP-C1⊂1), 受疏水和C—H…π相互作用驱动, SP-C1的N,N'-二甲基基团嵌入疏水空腔最深区域, 化学位移(δ)达-1.67, 另一端的甲基吡啶盐基团则裸露在重水中; 另一组为1∶2包结复合物(SP-C1⊂1.1), 此时SP-C1被完全封装于疏水空腔内, 其N,N'-二甲基基团化学位移(δ)为-1.37. 当加入量增至2.0 equiv.时, 氢谱信号以SP-C1⊂1.1为主导. 对于SP-C2体系, 向其重水溶液中加入1.0 equiv.分子杯1后, 核磁共振氢谱(见支持信息图S12)仅观测到SP-C2⊂1.1特征信号, 表明SP-C2的分子尺寸与胶囊1.1的疏水空腔高度匹配. 随着吡啶端烷基碳链的延长, 客体分子SP-Cn对分子杯1的二聚诱导行为呈现显著差异: SP-Cn⊂1.1主客体复合物中, N,N'-二甲基苯环质子(a、b和c)的化学位移先向高场移动后趋于稳定, 烷基吡啶盐质子(f、g和h)则先向低场偏移再转向高场. 这一动态变化表明, 短链烷基客体呈延展构象(SP-C2~SP-C4), 经向空腔中心压缩构象(SP-C5), 协同疏水作用与C—H…π作用驱动主体二聚; 当碳链超过临界长度时(SP-C5), 更长的烷基链(末端甲基的化学位移从高场移向低场)通过折叠“J”型构象适配空腔, 从而加强主体二聚驱动力[31](如图3所示). 其中, SP-C10的分子体积恰好对应主体二聚形成胶囊1.1的有效封装阈值, 而SP-C11和SP-C12因体积超出阈值, 无法诱导主体完成二聚封装(见支持信息图S13). 该现象揭示了客体构象自适应对主体二聚过程的层级调控机制, 即通过烷基链构象调整(延展→压缩→折叠)动态匹配主体空腔尺寸, 进而实现非共价分子胶囊的组装.
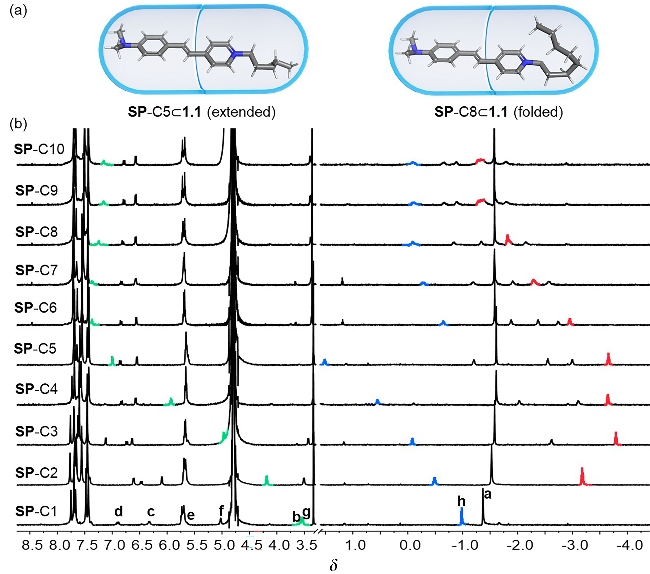
图3 (a) SP-C5和SP-C8在胶囊1.1中的构象示意图; (b) SP-Cn (n=1~10)在胶囊1.1中的部分1H NMR谱图(600 MHz, D2O, 298 K), 红色为烷基链上甲基Figure 3 (a) Conformational schematic illustration of SP-C5 and SP-C8 in Capsule 1.1; (b) Partial 1H NMR spectra of SP-Cn (n=1~10) in Capsule 1.1 (600 MHz, D2O, 298 K), red colors indicate methyl groups for alkyl chains |
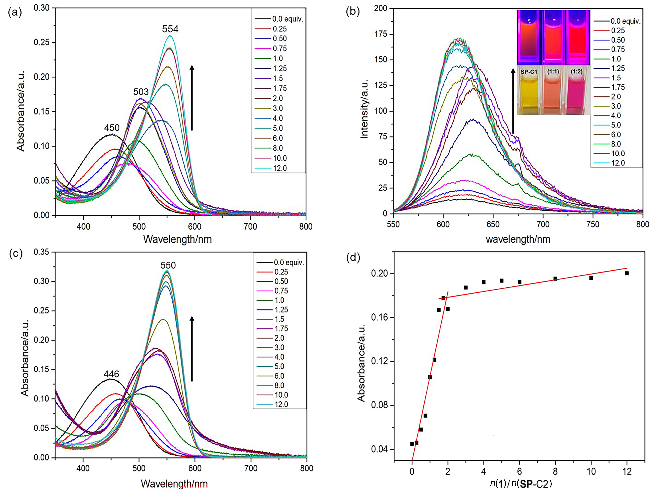
向SP-C1的水溶液中加入不同物质的量的分子杯1, 其紫外-可见吸收(UV-Vis)光谱表现出特征变化(图4a). SP-C1在450 nm处的初始吸收峰经历“降低→上升并红移至503 nm→再次降低→上升并红移至554 nm”的演变过程, 该现象与核磁共振结构形成互证, 反映出三个主客体作用阶段: 第一阶段为游离SP-C1和1∶1包结复合物(SP-C1⊂1)共存; 第二阶段过渡至1∶1包结复合物与1∶2包结复合物(SP-C1⊂1.1)共存; 第三阶段则以1∶2包结复合物为主. 在上述三种组装状态下, 体系于可见光范围内呈现出显著的颜色差异(图4b). SP-C1与分子杯1的荧光光谱分析(图4b)显示, 加入分子杯1后, SP-C1在620 nm处的发射强度显著增强12倍, 同时伴随发射波长红移. 这归因于胶囊限域环境对SP-C1分子内吡啶基与烯烃连接苯环相对旋转的抑制作用, 类似于聚集诱导发光(AIE)效应[32-33]. SP-C2的紫外-可见吸收光谱呈现相似的变化趋势(图4c). 相较于SP-C1, SP-C2与分子杯1形成的主客体复合物SP-C2⊂1.1在紫外光谱滴定中表现出典型的1∶2包结特征(图4d). 通过等温滴定量热法(ITC)测定SP-C2⊂1.1在水溶液中的结合常数, 获得逐级结合常数Ka1=9.97×108 L•mol−1和Ka2=1.48×105 L•mol−1(见支持信息图S15). 该结果表明, SP-C2与分子杯1之间存在强亲和力的两步组装过程.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图4 SP-C1的水溶液(2.5 μmol/L)在不同物质的量1存在下的紫外吸收光谱(a)和荧光光谱图(b), 激发波长为450 nm; 插图为SP-C1⊂1在日光或405 nm光下的图像; (c) SP-C2的水溶液(2.5 μmol/L)在不同物质的量1存在下的紫外吸收光谱; (d)在515 nm处吸光度与n(1)/n(SP-C2)物质的量比的关系图Figure 4 The absorption spectra (a) and fluorescent spectra (b) of SP-C1 in water in the presence of incremental amount of 1; Inset: photograph of SP-C1⊂1 under daylight or 405 nm light (c=2.5 μmol/L, λex=450 nm); (c) The absorption spectra of SP-C2 in water in the presence of incremental amount of 1; (d) The absorbance at 515 nm versus molar ratio of n(1)/n(SP-C2) |
3 结论
本工作通过在2-氨基苯并咪唑基间苯二酚分子杯 1的下缘引入羧酸钠水溶性基团, 显著提升了其在水相中的溶解性能, 并结合¹H NMR、¹³C NMR及HRMS完成了结构表征. 基于该主体分子, 系统研究了其与对称型直链烷烃及非对称型直链烷基苯乙烯类半菁荧光染料分子(SP-Cn)的主客体自组装行为. 1H NMR和2D NOESY表明, 对称型直链烷烃(如正十二烷至正二十烷)可通过从分子两端向中间压缩的构象调整策略以适配空腔尺寸, 协同疏水作用诱导分子杯1发生二聚, 形成具有动态限域结构的胶囊1.1. 对于非对称型烷基SP-Cn系列染料分子, 其柔性烷基链在胶囊疏水空腔内被压缩, 形成独特的“J”型折叠构象, 同样印证了客体构象自适应对主体二聚的驱动机制. 光谱学研究进一步表明, SP-C1分子通过疏水作用、阳离子…π相互作用及C—H…π相互作用被锚定在胶囊空腔内, 其荧光发射强度显著增强12倍, 并表现出高亲和力(结合常数Ka12达1013 L2•mol−2). 这种基于主客体相互作用的荧光增强效应, 为构建超分子荧光探针及逻辑门器件提供了新策略.
本工作突破了传统分子胶囊依赖主体预设互补位点的局限, 建立了“客体构象变化→非共价键协同作用→主体动态二聚”的超分子组装新机制. 未来研究将聚焦于利用该主客体体系构建荧光传感器阵列, 拓展其在DNA结构识别与生物分子检测领域的应用.
(Cheng, B.)