

1 引言
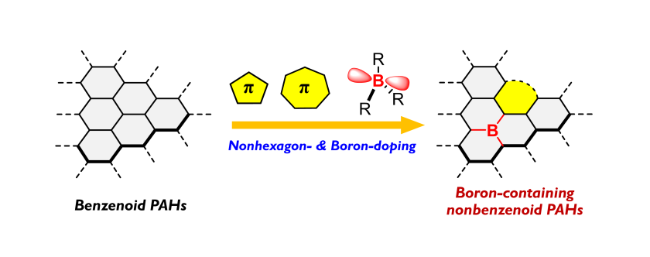
2 硼氮共杂非苯稠环芳烃

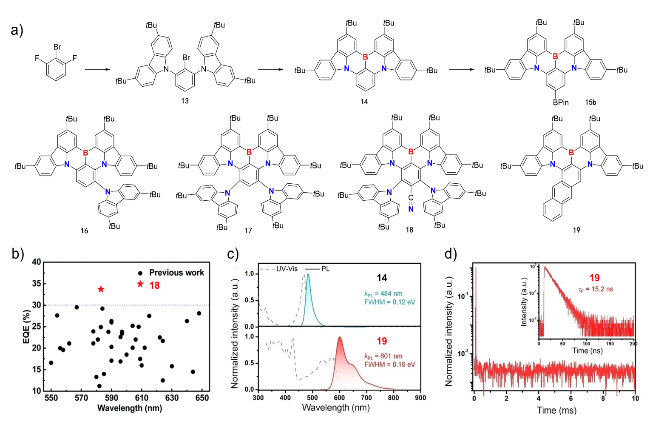
图3 (a)硼氮共杂非苯稠环芳烃14及其硼酯15b的合成路线和四例多重共振分子14~19的化学结构; (b)发射峰为550至650 nm的TADF-OLED的EQEmax总结[84]; (c)甲苯中14和19的紫外吸收和荧光光谱; (d)甲苯中19的瞬态衰减曲线[89]Figure 3 (a) Synthetic route of B/N co-doped nonbenzenoid PAHs 14 and 15b, and chemical structures of B/N co-doped nonbenzenoid PAHs 14~19; (b) EQEmax summary of TADF-OLEDs with an emission peak from 550 to 650 nm[84], Copyright 2021 Royal Society of Chemistry; (c) UV/Vis absorption and fluorescence spectra of 14 and 19 in toluene; (d) Transient PL decay curves of 19 in toluene solution[89]. Copyright 2024 WILEY-VCH |

图4 (a, b)硼氮共杂非苯稠环芳烃21、22和25的合成路线; (c, d)甲苯中21和22的紫外吸收光谱、荧光光谱、圆二色谱图和圆偏振发光光谱图[90]; (e)全碳和B/X掺杂茚并芴的双自由基指数和单-三线态能隙[91]Figure 4 (a, b) Synthetic routes of B/N co-doped nonbenzenoid PAHs 21, 22 and 25; (c) UV/Vis absorption and fluorescence spectra, (d) CD and CPL spectra of 21 and 22[90]; (e) Diradical characters and singlet-triplet energy gaps of s-indacene-based all-carbon and B/X-doped heterocyclic PAHs[91]. Copyright 2023, 2024 WILEY-VCH |
3 硼氧/硼硫共杂非苯稠环芳烃
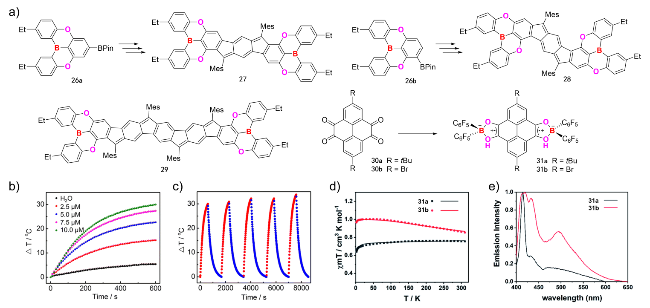
图5 (a)硼氧共杂非苯稠环芳烃27、28和31的合成路线、以及29的化学结构; (b)不同浓度的29在激光照射下的温度变化[94]; (c) 29在经历5个激光开关循环后的加热与冷却曲线[94]; (d) 31的超导量子干涉仪测试图[95]; (e)四氢呋喃中31的荧光光谱图[95]Figure 5 (a) Synthetic routes of B/O co-doped nonbenzenoid PAHs 27, 28 and 31, and chemical structure of 29; (b) Temperature variations of various concentrations of 29 under laser irradiation[94]; (c) The heating and cooling curves of 29 upon 5 cycles of laser on and off[94]; (d) The SQUID measurements of the powder 31[95]; (e) Fluorescence spectra of 31[95]. Copyright 2021, 2023 Royal Society of Chemistry |

4 纯硼杂非苯稠环芳烃
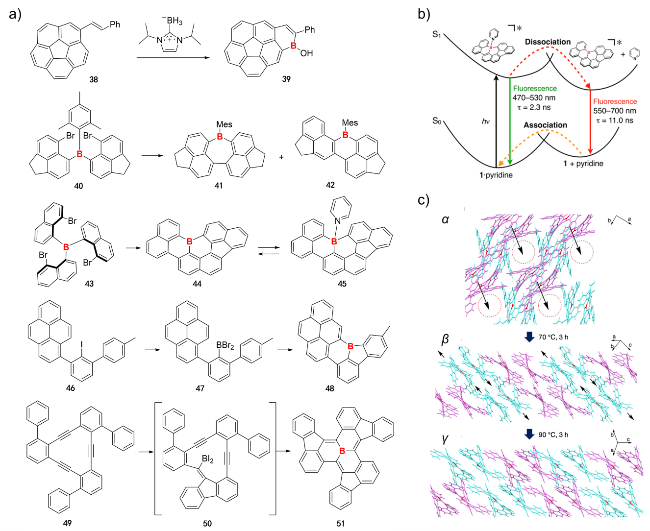
图7 (a)硼杂非苯稠环芳烃39、41、42、44、45、48和51的合成路线; (b) 45的光致解离机理[101]; (c) 51-α向51-γ转换的机制[104]Figure 7 (a) Synthetic routes of B-doped nonbenzenoid PAHs 39, 41, 42, 44, 45, 48 and 51; (b) A plausible energy diagram for the photodissociation of 45[101]; (c) Estimated transformation mechanism from 51-α to 51-γ[104]. Copyright 2014, 2024 American Chemical Society |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}