


1 引言
2 MOF复合物的类型
2.1 客体材料的类型
表1 不同类型复合策略在结构调控、电化学性能和合成工艺可扩展性等方面的比较Table 1 Comparison of different types of composite strategies in structural regulation, electrochemical performance, and scalability of synthesis processes |
| MOF复合物类型 | 结构调控的核心策略 | 电化学性能提升机制 | 合成工艺 |
|---|---|---|---|
| 金属氧化物@MOFs | 通过金属价态调控与异质界面设计优化电子结构 | 法拉第赝电容与协同电子传导 | 溶剂热、电化学沉积 |
| 无机碳材料@MOFs | 以MOF为前驱体, 通过热解与孔结构设计实现碳基功能化 | 双电层电容与表面赝电容 | 高温热解、模板法中空化 |
| 聚合物@MOFs | 引入多种官能团, 实现氧化还原活性、离子传导和 界面稳定化的协同 | 增强柔性、离子通透性与稳定性 | 原位聚合、一锅法 |
2.1.1 金属氧化物@MOFs
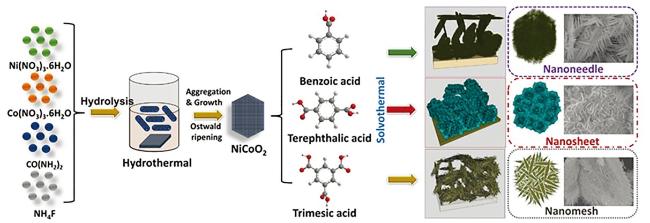
2.1.2 无机碳材料@MOFs
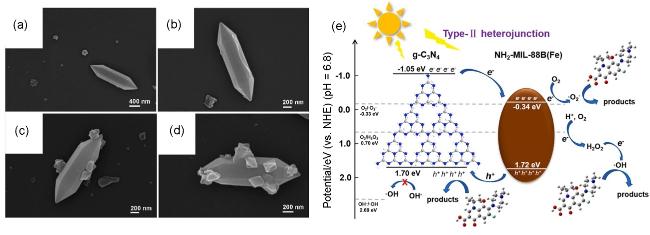
图2 (a, b) NH2-MIL-88B(Fe)和(c, d) g-C3N4/NH2-MIL-88B(Fe)的FESEM[22]及(e) g-C3N4/NH2-MIL-88B(Fe)复合材料在可见光照射下对OFL光降解过程的示意图[22]Figure 2 FESEM images of (a, b) NH2-MIL-88B(Fe) and (c, d) g-C3N4/NH2-MIL-88B(Fe)[22], and (e) schematic diagram showing OFL photodegradation on the g-C3N4/NH2-MIL-88B(Fe) composite under visible light irradiation[22] |
2.1.3 聚合物@MOFs
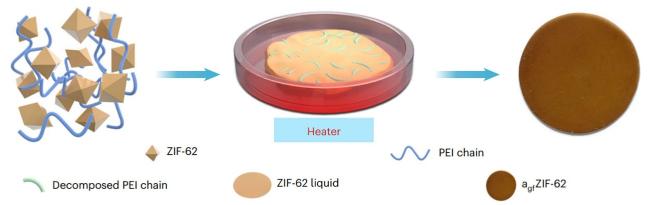
2.2 MOF复合材料的维度特征
2.2.1 0D MOF
2.2.2 1D MOF
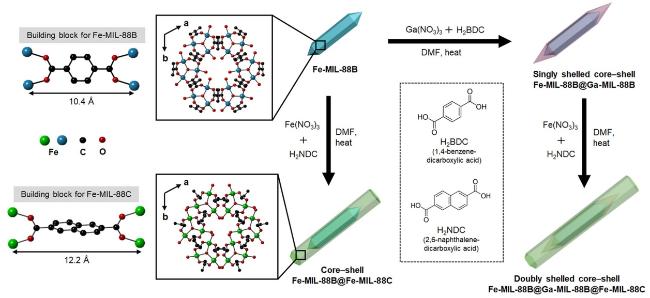
图4 通过尖端至中心各向异性MOF-on-MOF生长策略, 在晶格失配条件下制备核壳型杂化MOFs (Fe-MIL-88B@Fe-MIL-88C和Fe-MIL-88B@Ga-MIL-88B@Fe-MIL-88C)的示意图[30]Figure 4 Schematic representation for the production of core-shell-type hybrid MOFs (Fe-MIL-88B@Fe-MIL-88C and Fe-MIL-88B@Ga- MIL-88B@Fe-MIL-88C) via the tip-to-middle anisotropic MOF-On-MOF Growth with Mismatched Cell Lattices[30] |
2.2.3 2D MOF

2.2.4 3D MOF

3 MOF复合物在能源储存中的应用
3.1 MOF复合物应用于超级电容器
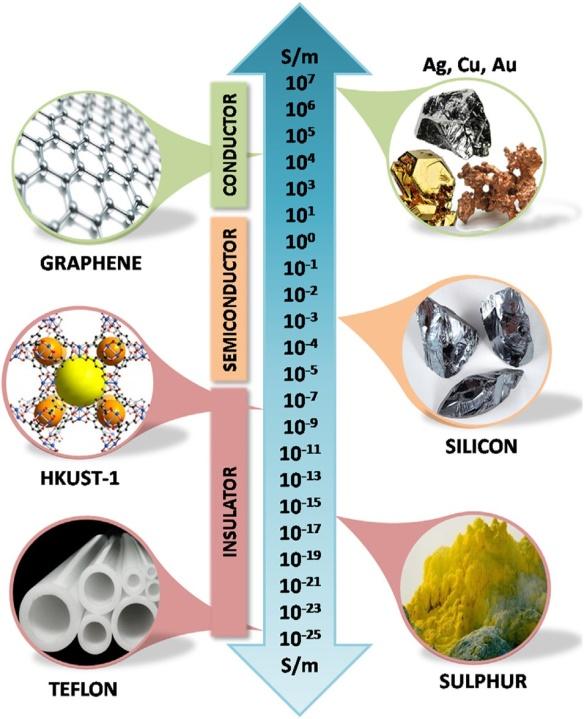
3.1.1 超级电容器的分类
3.1.2 石墨烯及其衍生物@MOFs
3.1.3 金属氧化物@MOFs
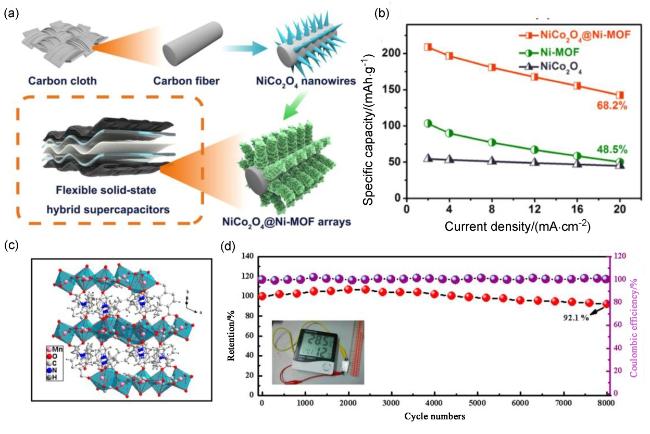
图8 (a)碳布上NiCo2O4@Ni-MOF核/壳杂化阵列的合成方法示意图[47]、(b) NiCo2O4@Ni-MOF在不同放电电流密度下的比容量[47]、(c) Mn-MOF的线框视图[48]及(d) K0.5Mn2O4@Mn-MOF-8在恒定电流密度2 A•g-1下的循环性能[48]Figure 8 (a) Schematic illustration of the synthetic approach for the NiCo2O4@Ni-MOF core/shell hybrid arrays on carbon cloth[47], (b) specific capacity of NiCo2O4@Ni-MOF at various discharge current densities[47], (c) wireframe view of the Mn-MOF[48], and (d) cycling performance of K0.5Mn2O4@Mn-MOF-8 at constant current density of 2 A•g-1[48] |
3.1.4 聚合物@MOFs
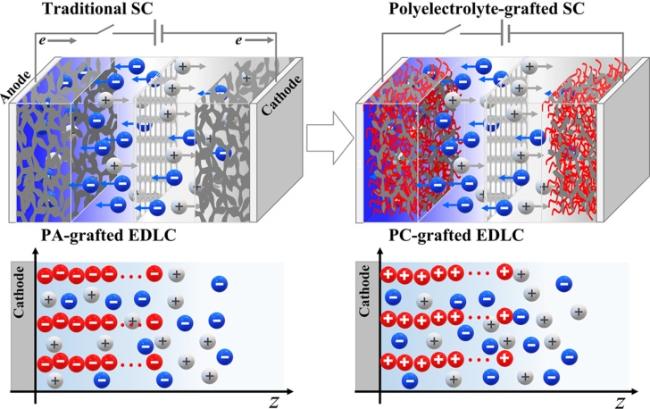
3.2 MOF复合物应用于锂离子电池
表2 MOF复合物在锂离子电池中的应用Table 2 Application of MOF composites in lithium-ion batteries |
| MOF复合物 | 客体材料 | 应用 | 比容量 | 容量保持率 | 文献 |
|---|---|---|---|---|---|
| PPHK | 聚吡咯 | 锂离子电池阳极 | 147.2 mAh•g-1 at 1 C | 86.7% after 800 cycles | [59] |
| Si/CNTs@ZIF-800N | 硅, 碳纳米管 | 锂离子电池阳极 | 1223 mAh•g-1 at 0.1 A•g-1 | 98.4% after 100 cycles | [60] |
| Porous ZnCo2O4/C | 碳纤维 | 锂离子电池阳极 | 1707 mAh•g-1 at 0.1 A•g-1 | 67.1% after 100 cycles | [61] |
| Co-TCPP MOF/rGO | 还原氧化石墨烯 | 锂离子电池阳极 | 2317 mAh•g-1 at 0.1 A•g-1 | ≈45.3% after 100 cycles | [62] |
| PPy@Cu-MOFs | 聚吡咯 | 锂离子电池隔膜 | 150.2 mAh•g-1 at 0.5 C | ≈100% after 200 cycles | [32] |
| PP@MIL-101-COOH | 聚偏二氟乙烯 | 锂离子电池隔膜 | — | 95.9% after 150 cycles | [55] |
| PH-SSE | 聚偏二氟乙烯 | 锂离子电池电解质 | — | 85.1% after 300 cycles | [54] |
| BMOF@HF/H-ZIF-8 | 聚丙烯腈中空纤维 | 锂离子电池电解质 | — | 90.45% after 200 cycles | [58] |
3.2.1 硅基材料@MOFs
3.2.2 MXene@MOFs
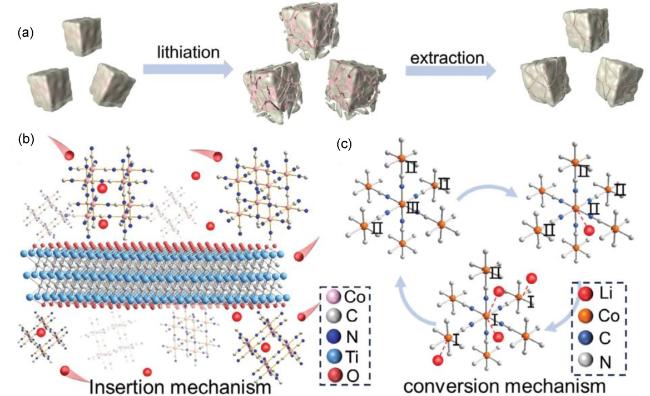
图10 (a) Co-PBA/MXene在循环过程中Li+插入和提取时核壳状结构发生变化的示意图[61]、(b, c) Co-PBA/MXene的储能机制图[61]Figure 10 (a) Schematic illustration of the core-shell structural changes in Co-PBA/MXene during the insertion and extraction of Li+ in the cycling process[61], (b, c) diagram of the lithium storage mechanism of Co-PBA/MXene[61] |
3.2.3 聚合物@MOFs
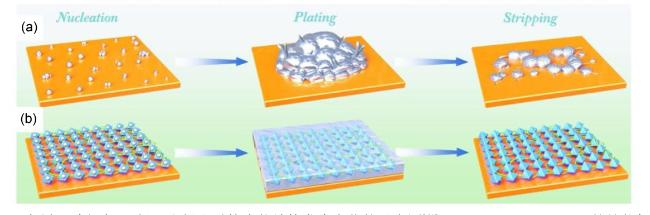
3.3 MOF复合物应用于锂硫电池
表3 MOF复合物应用于锂硫电池Table 3 Application of MOF composites in lithium sulfur batteries |
3.3.1 MOF复合物作为“硫宿主”
3.3.2 MOF复合物作为隔膜材料
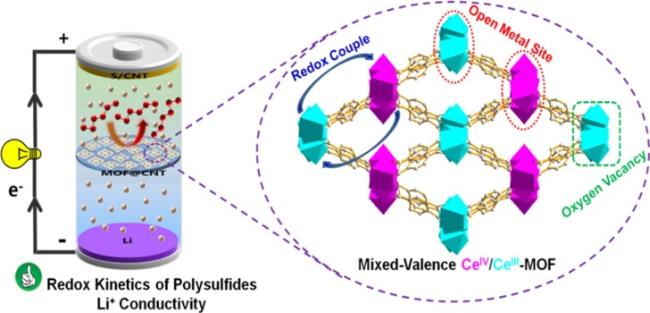
3.4 MOF复合物应用于水系锌离子电池
表4 MOF复合物在水系锌离子电池中的应用Table 4 Application of MOF composites in aqueous zinc ion battery |
| MOF复合物 | 客体材料 | 应用 | 比容量 | 容量保持率 | 文献. |
|---|---|---|---|---|---|
| MIL-88B(V)@rGO | 氧化石墨烯 | 锌离子电池阴极 | 480 mAh•g-1 at 0.05 A•g-1 | 80.3% after 400 cycles | [84] |
| Mn-MOF/CNT | 碳纳米管 | 锌离子电池阴极 | 260 mAh•g-1 at 0.05 A•g-1 | ≈100% after 900 cycles | [85] |
| V-MOF@graphene | 石墨烯 | 锌离子电池阴极 | 342 mAh•g-1 at 0.1A•g-1 | ≈89% after 100 cycles | [86] |
| MXene/Cu-THBQ | MXene | 锌离子电池阳极 | 235.4 mAh•g-1 at 0.2 A•g-1 | 98.7% after 400 cycles | [87] |
3.4.1 MOF复合物作为正极材料
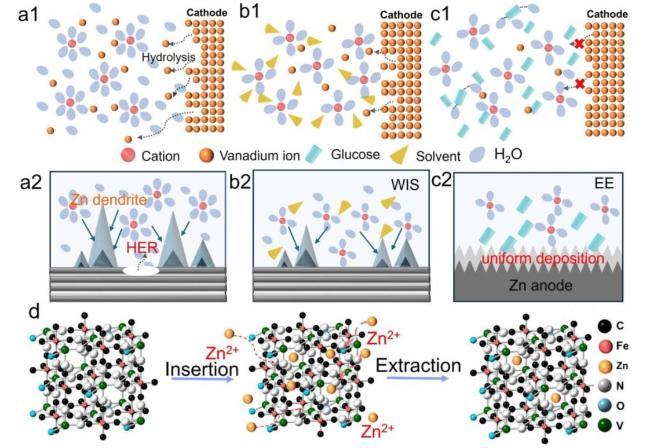
图13 (a1~c1) V-PBA正极在3 mol/L Zn(OTf)2、盐水和共晶电解质中的溶解行为示意图[89], (a2~c2)锌枝晶在3 mol/L Zn(OTf)2、盐水和共晶电解质中的生成过程示意图[89]及(d) V-PBA储存Zn2+机制的示意图[89]Figure 13 (a1~c1) Schematic diagram of the dissolution behavior of the V-PBA cathode in 3 mol/L Zn(OTf)2, saline, and eutectic electrolytes[89], (a2~c2) schematic diagram of the formation process of zinc dendrites in 3 mol/L Zn(OTf)2, saline, and eutectic electrolytes[[89], and (d) schematic diagram of the mechanism of Zn2+ storage in V-PBA[89] |
3.4.2 MOF复合物作为负极涂层

4 MOF复合物在吸附中的应用
4.1 吸附金属离子
4.1.1 U(Ⅵ)
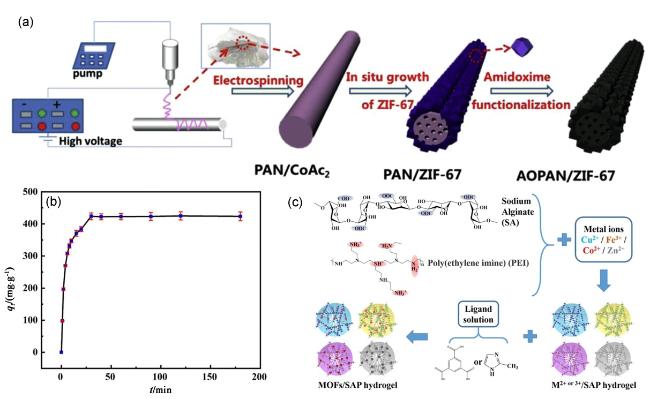
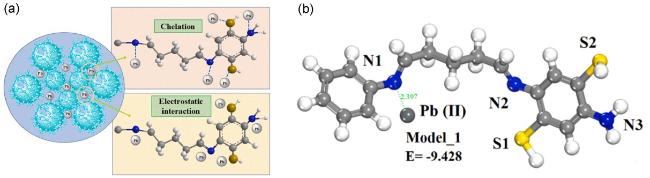
4.1.2 Pb(Ⅱ)
4.2 气体的吸附
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
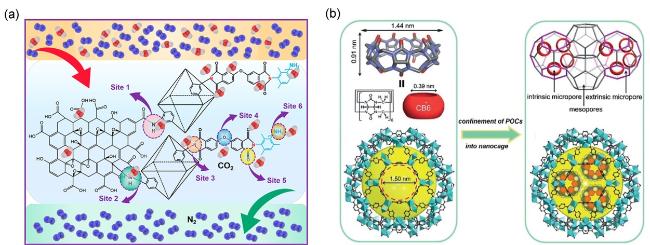
图17 (a) PI-UiO/GOs的交联、缩合和亲核反应机理以及吸附相互作用位点[101]及(b)通过将CB6结合到铬基MIL-101中来创建功能性杂化材料的概念图[104]Figure 17 (a) Cross-linking, condensation, and nucleophilic reaction mechanism of PI-UiO/GOs and adsorption interaction sites[101] and (b) conceptual diagram of functional hybrid materials by incorporating CB6 into chromium based MIL-101[104] |