

1 引言
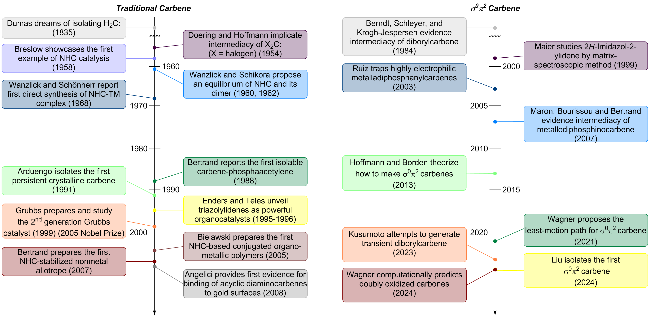
卡宾(R2C:)是一类二价碳化合物, 其中心碳原子具有六电子结构特征. 其历史可以追溯到1835年, 当时Dumas[4]声称自己梦见分离了H2C:物种, 即最简单的卡宾——二氢卡宾(如图1). 直到1954年, Doering和Hoffmann[5]才提供了二氯卡宾作为反应中间体的证据, 揭示了卡宾这一物种存在的可能性. 1958年, Breslow[6]展示了噻唑盐催化安息香缩合(benzoin condensation)的实例. 随后(1960年和1962年), Wanzlick和Schikora[7]提出了氮杂环卡宾(N-heterocyclic carbene, NHC)与其二聚体之间存在的一种平衡(Wanzlick equilibrium). 到1968年, Wanzlick和Schönherr[8]报道了第一例直接合成的NHC-过渡金属配合物. 然而, 直到1988年, Bertrand等[9]才成功分离了首例稳定的磷基硅基卡宾. 三年后, Arduengo等[10]报道了首例NHC卡宾. 1995~1996年, Enders和Teles等[11]揭示了三氮咪唑衍生的NHC作为有机催化剂的潜力. 值得一提的是, 1999年Grubbs等[12]利用NHC发展出第二代Grubbs催化剂. 这些开创性发现表明, 卡宾, 这一传统上被认为是瞬时存在且不稳定的化合物, 实际上可以像普通化学试剂一样被使用, 并具有广阔的应用前景. 因此, 稳定的卡宾现已成为合成催化[13]、材料科学[14]和药物化学[15]等许多领域的宝贵工具.
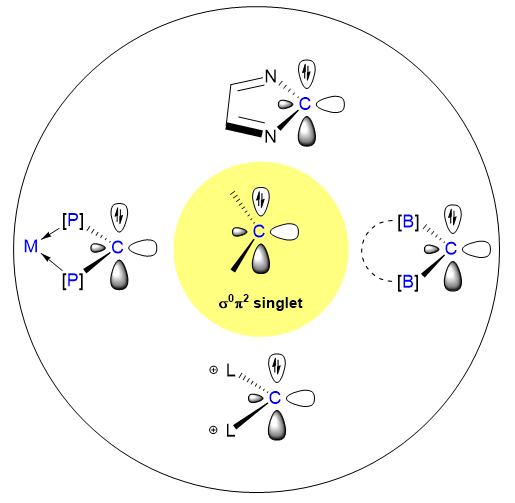
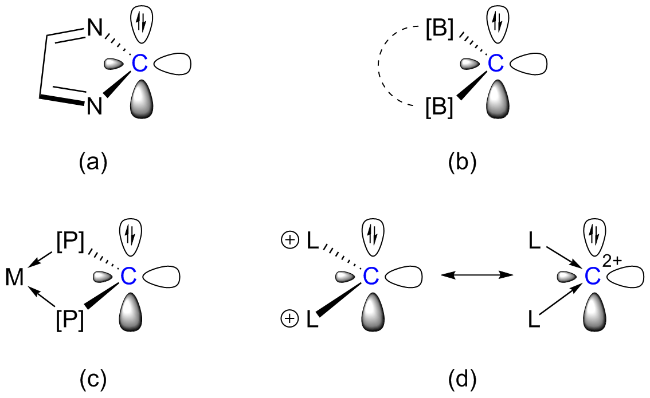
卡宾的前线轨道可以系统性地分为σ轨道(平面内)和π轨道(平面外)[16]. 通过对卡宾的电子构型进行分析, 卡宾可以被分为四类. 两个非键电子可以自旋相同或相反分布在不同的轨道上, 即为σ1π1电子态. 根据这两个不同轨道上电子的自旋方向, 卡宾分别为σ1π1三线态[17]或σ1π1单线态(如图2). 与此相反, 卡宾的两个非键电子也可以成对分布在同一轨道上. 根据电子分布轨道的不同, 可以分为σ2π0单线态或σ0π2单线态(如图2). 通常情况下, σ轨道的能量比π轨道的能量低(σ轨道具有s轨道的杂化贡献, π轨道由纯p轨道组成), 使得目前分离得到的稳定卡宾几乎都是σ2π0单线态[16], 少数是σ1π1三线态[17]. 截至目前, 分离得到的“反转电子态(σ0π2)”卡宾仅有一例(详见下文). 值得一提的是, 两个电子以自旋相反的形式分别占据不同轨道形成的单线态σ1π1卡宾还未被科学家分离表征. 本综述将对反转电子态(σ0π2)卡宾的相关研究情况进行阐述和总结, 并展望这一领域未来的发展.
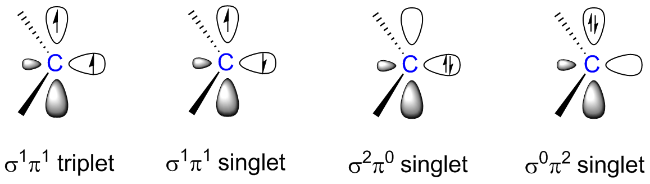
2 反转电子态(σ0π2)卡宾的相关研究情况
2.1 反转电子态卡宾的稳定策略
2013年, Hoffmann和Borden等[20]提出将卡宾碳嵌入包含低能量π轨道的环中, 有望合成电子基态为σ0π2的卡宾. 作者计算了相应结构不同电子构型的能量, 说明了环状二亚氨基卡宾基态电子构型为σ0π2.
对于二硼基卡宾的相关理论研究则更早. 1990年, Cioslowski等[21]计算了硼杂苯及其与一氧化碳、氮气加合物的电子结构; 随后, 在1997年, Karadakov, Ellis, Gerratt, Cooper和Raimondi[22]详细地研究了硼杂苯的电子结构. 1995年, Berndt等[23]研究了1,3-diboretane重排中的活性中间体, 二硼基卡宾. 作者计算了二硼基卡宾的电子构型, 证明了其基态为σ0π2. 2013年, Kassaee 等[24]利用密度泛函理论(DFT)计算对2,2,9,9-四甲基环壬-3,5,7-三烯卡宾(2,2,9,9-tetramethylcyclonona-3,5,7- trienylidene)及其杂环类似物进行了研究. 作者研究了不同杂原子以及环系中的双键对卡宾碳中心电子构型的影响, 推断当π受体/σ给体(如BH, AlH, SiH2等基团)与卡宾碳原子相连时, 卡宾的基态电子构型将呈现σ0π2. 2015年, Akbari等[25]对2,5-二(卤硼基)环戊卡宾(2,5-bis(halobora)cyclopentenylidene)及其重主族元素(Si, Ge, Sn, Pb)类似物进行了理论研究, 说明了硼基作为π电子受体的潜在优势.
在2005年, Uría等[26]通过理论计算研究了金属配位的环状二磷基卡宾体系. 作者探究了[Mn(CO)4(PH2)2C:]+卡宾的结构和电子性质, 并与[Ru(CNH)4(PH2)2C:]2+卡宾进行了比较, 说明了此类卡宾基态电子态呈现σ0π2的理论可能性.
在更近的2024年, Wagner[27]提出双阳离子卡本可以看作σ0π2卡宾. 卡本(carbone)或碳二卡宾(carbodicarbene, CDC), 形式上可看作两个中性配体直接配位在一个碳原子上, 其一般结构为L→C←L. 这种中性的二配位螯合的碳原子具有两对孤对电子, 具有很强的配位能力, 这使得卡本及其相关配合物在催化等方面展现出独特的应用. 移除卡本碳原子的两个电子则得到双阳离子卡本. 双阳离子卡本结构中存在配体向σ轨道提供电子和接受π轨道电子(负超共轭)的两种作用. 后一种相互作用的重要性表明, 通过从碳二磷烷(carbodiphosphoranes)移除两个电子形成双阳离子卡本, 可以实现类似的电子结构. 作者优化了几种双阳离子各自的单线态和三线态的结构和能量, 并在可能的情况下使用高精度的G4MP2方法进一步计算了单线态-三线态能隙. 研究表明, 这种策略有希望得到σ0π2卡宾. 但是, 相关的实验研究还未见发表, 因此这一体系不会在后文详细讨论.
2.2 环状二亚氨基卡宾的相关研究进展
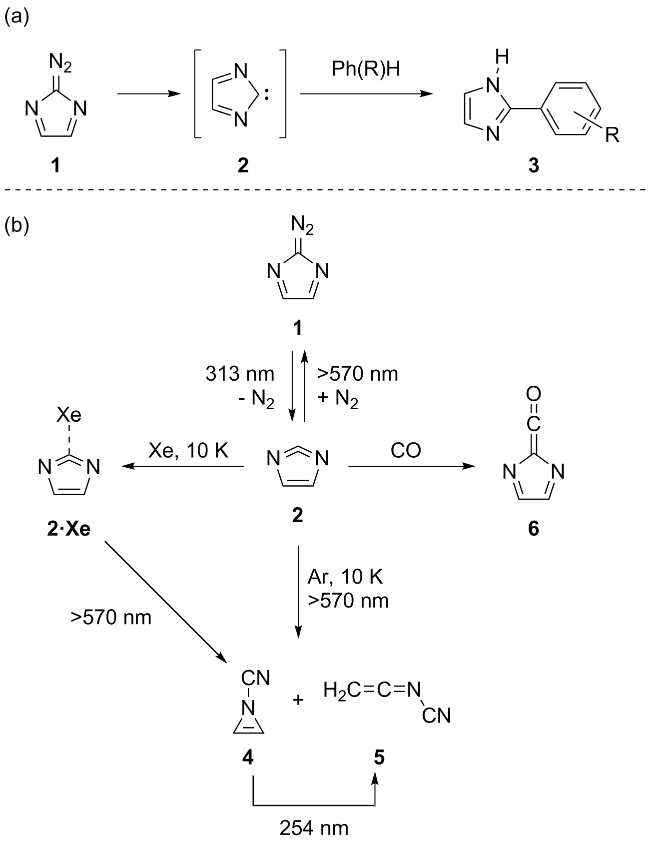
1999年, Maier等[28]对环状二亚胺基卡宾2 (2H-imidazol-2-ylidene)进行了基质光谱学研究. 作者在10 K低温下对基质中的前体1 (2-diazo-2H-imidazole)进行光解, 成功制备了卡宾2, 并用苯的衍生物进行了捕获, 生成化合物3(图5(a)). 随后, 作者在基质中研究了2的反应性, 包括与稀有气体(Xe)、一氧化碳等(图5(b)). 此外, 作者还通过理论计算对环状二亚胺基卡宾的结构与电子构型等进行了深入探究. 结果表明, 其基态为单线态, 并具有C2v对称性. 在2的结构中, N—C—N键角较大(约140°), N—Ccarbene键长较短(约125 pm, 结构中另一N—C键键长约为145 pm), C—N—C键角较小(约87°). 这些特征表明其具有累积二烯结构而非“典型的卡宾结构”.
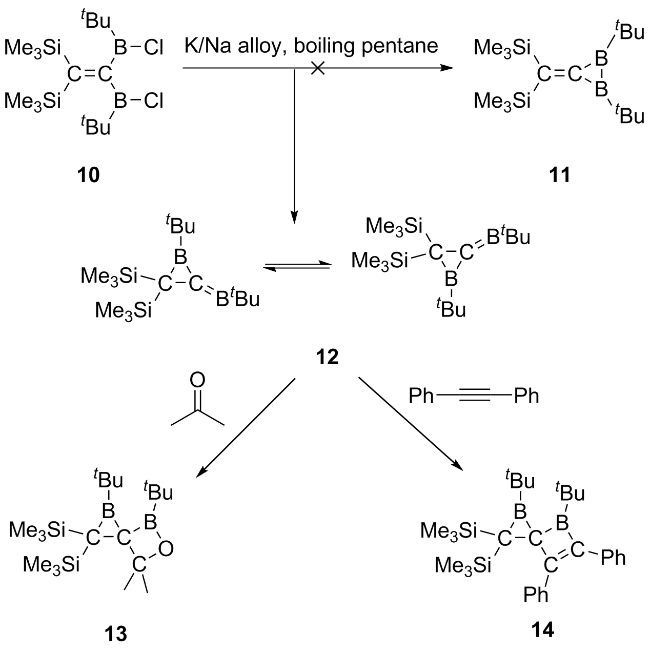
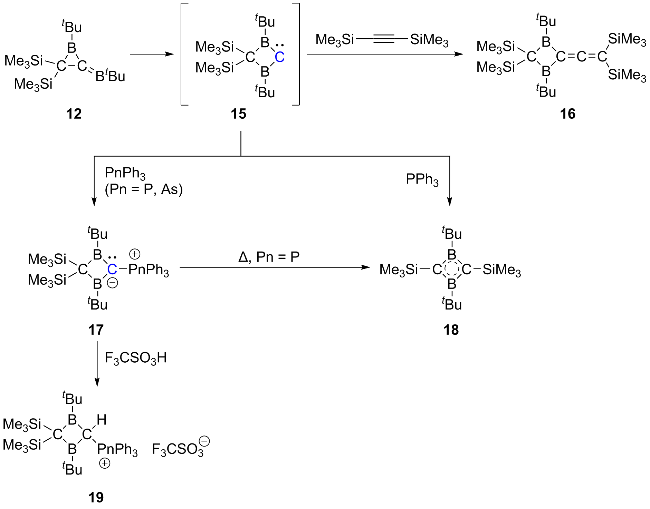
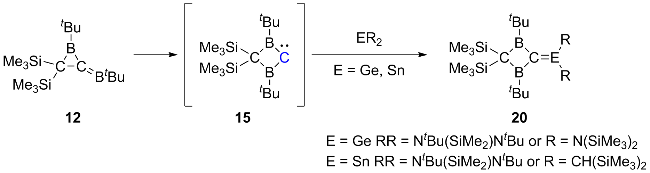
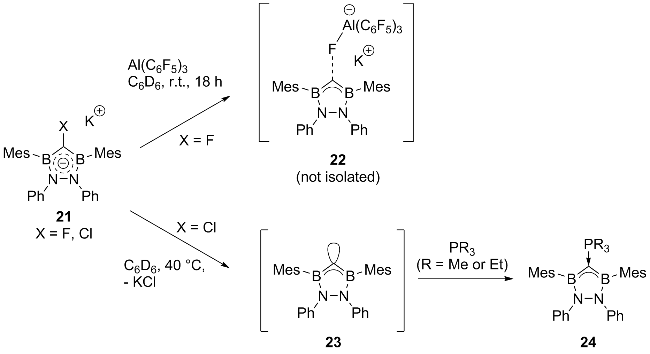
2.3 二硼基卡宾的相关研究进展
2.4 金属配位的环状二磷基卡宾的相关研究进展
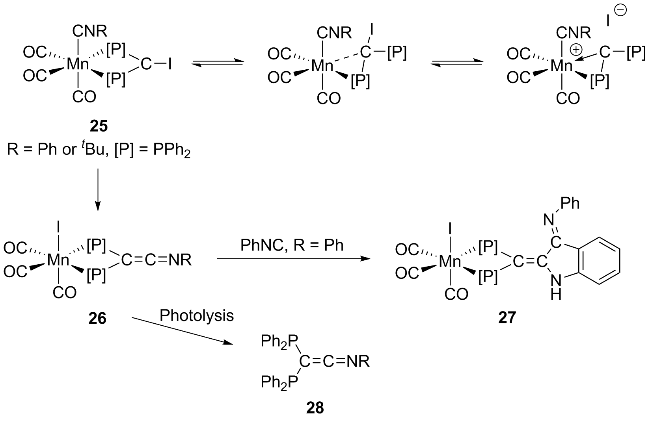
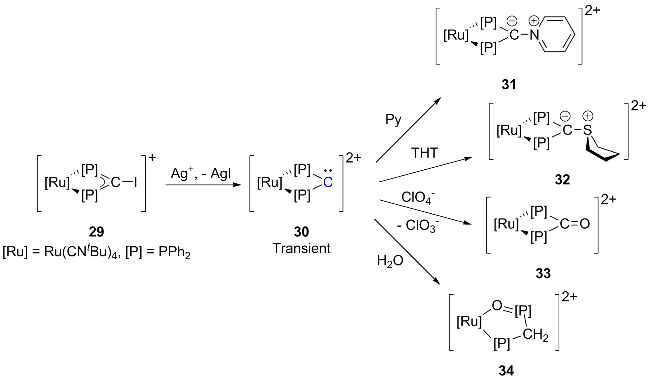
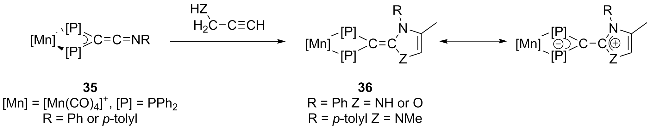

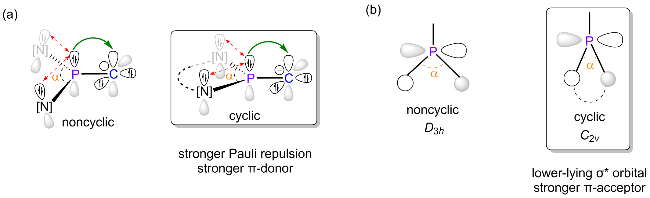
图16 (a)非环状与环状二氨基磷取代基的π供体能力对比; (b)平面磷基取代基的电子结构前沿分子轨道图(基于理想化D3h和C2v点群)Figure 16 (a) Comparison of the π-donating ability for noncyclic and cyclic diaminophosphino substituents; (b) Frontier molecular orbital diagrams depicting the electronic structure of planar phosphine substituents with the idealized D3h and C2v point groups |

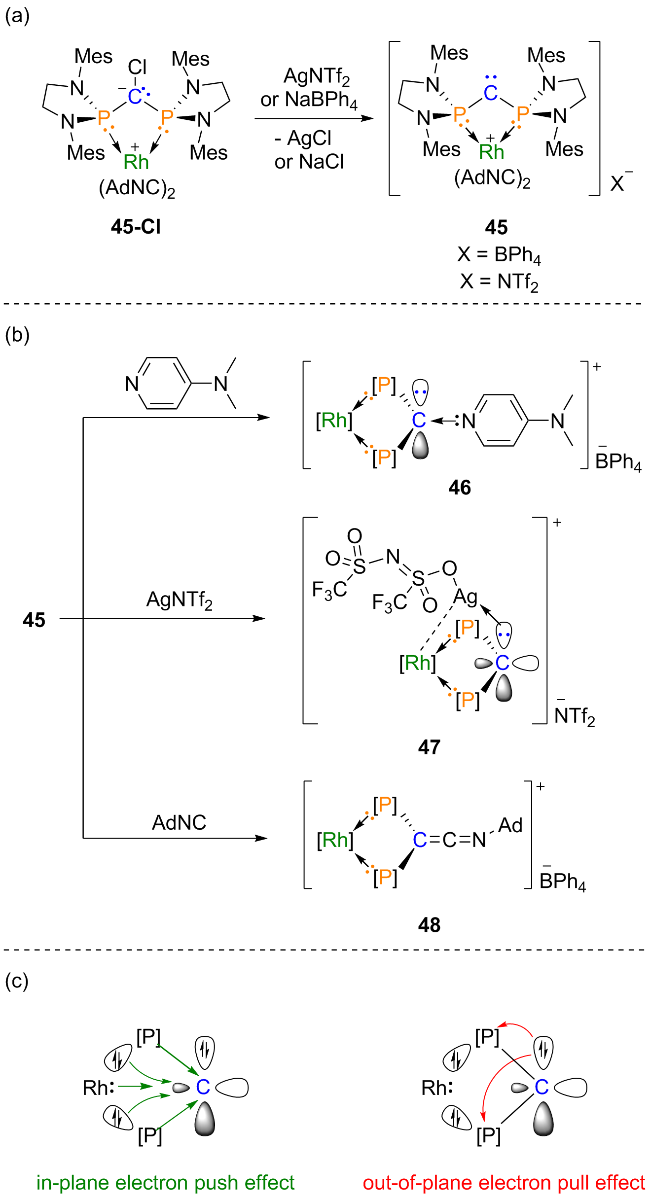
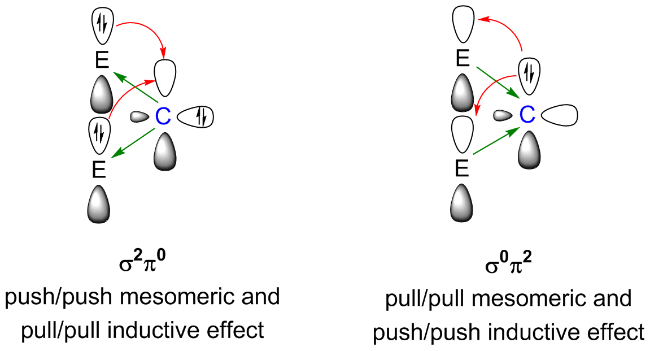
通过理论计算分析, 研究者深入揭示了该体系的电子调控机制: 一方面, 磷基配体通过负超共轭效应接受卡宾碳中心的π电子(图18(c)); 另一方面, 金属铑中心及配体向卡宾碳的σ空轨道提供电子. 这种推拉协同的电子相互作用有效稳定了卡宾碳的σ0π2电子态. 该研究不仅为新型卡宾体系的构建提供了创新策略, 同时为小分子活化和卡宾配位化学开辟了新的研究方向.
该例研究发现, 与传统的σ2π0卡宾相比, 反转电子态(σ0π2)卡宾不仅在电子构型上不同, 也在核磁共振谱学性质和反应性等方面有显著差异. 在核磁共振碳谱表征中, 卡宾45中卡宾碳谱信号出现在δ -30.9(化合物45, X=BPh4)和δ -32.3(化合物45, X=NTf2)[48], 明显偏离了与之类似的磷杂环卡宾(P-heterocyclic carbenes, PHCs)中卡宾碳原子的碳谱信号(大于δ 180)[49]和被广泛研究的氮杂环卡宾(N-heterocyclic carbenes, NHCs)中的信号(通常大于δ 185)[50]. 在反应性上, 卡宾45也表现出了不同于传统σ2π0卡宾的更强亲电性(如图18中接受4-二甲氨基吡啶(DMAP)配位生成化合物46)[48].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
换一个分析角度, 锡炔49含有两个异核的双亲性元素中心: 碳和锡. 由于Pauli排斥, 碳和锡不会同时将孤对电子置于平行排列. 在49中, 锡的孤对电子位于平面内, 碳的孤对电子则位于面外. 基于此, 锡炔的稳定合成意味着, 通过精准的电子结构调控, 开环构型反转电子态σ0π2卡宾的合成与分离在实验化学层面具有可行性.
3 总结和展望
尽管反转电子态(σ0π2)卡宾的研究已取得初步进展, 其发展仍面临诸多挑战与机遇. 目前文献报道的四个典型体系中, 仅金属配位的环状二磷基卡宾成功实现了稳定分离, 其余三类体系尚待突破. 对这些体系的深入探索将有望为σ0π2卡宾的稳定化策略提供新思路.
值得关注的是, 将反转电子态概念延伸至卡宾类似物的构建, 可能为主族元素化学开辟新方向. 这类体系的结构特征、成键模式及反应特性研究, 有望突破传统主族元素化学的认知框架. 此外, 基态为σ1π1的单线态卡宾至今仍未实现稳定分离, 这一科学难题持续激发着研究者的探索热情. 随着先进合成技术与理论计算方法的协同发展, 该体系的突破性进展将为卡宾化学注入新的活力, 深化对这类特殊物种的构效关系理解.
(Cheng, B.)