

1 引言
聚酰亚胺(Polyimide, PI)薄膜因其优异的耐高温、高绝缘及化学稳定性等性能被广泛应用于光电器件、生物传感、半导体芯片封装等战略新兴领域, 素有“黄金薄膜”之称[1-9]. 随着纳米技术的迅猛发展, 光电设备向轻薄化、小型化及高集成化方向演化; 借助光刻技术, 人们已成功将纳米级厚度的PI超薄膜用于电子封装中的介电材料[10]. 然而, 当高分子薄膜厚度降低至纳米尺度, 尤其是接近高分子链尺寸时, 其性能会偏离本体性质[11-15], 表现出纳米受限效应. 例如, 厚度在100 nm以下的聚苯乙烯薄膜具有比本体更低的玻璃化转变温度(Tg)[16-17]、更快松弛速率[18-19]和更低黏度[20-21], 致使材料易于发生去润湿等界面失稳现象[22-24]; 此外, 纳米级的聚甲基丙烯酸薄膜的热膨胀系数随着膜厚的降低而增大[25-26], 使薄膜的尺寸稳定性降低而造成材料失效. 这些都严重地限制了材料的实际应用. 因此, 研究和阐明受限状态下PI超薄膜的分子松弛行为, 对优化微纳精密器件的结构设计和性能调控具有重要意义.
过去三十年间, 纳米聚合物薄膜松弛动力学受到广泛关注; 然其研究主要集中于聚苯乙烯[16-18]、聚甲基丙烯酸甲酯[25-27]、聚醋酸乙烯酯[28]等为代表的柔性高分子; 而对PI等刚性高分子体系的研究甚少. 另一方面, Wang等[29-30]发现聚咔唑类(如PCDTBT)等刚性共轭高分子的Tg随膜厚的降低而减小; Bradley等[31]却发现聚9,9-二辛基芴的Tg随膜厚降低出现先升高后降低的非单调趋势; 而Fytas等[32]发现了纳米级聚酰亚胺薄膜的弹性模量不依赖于厚度, 这说明刚性高分子具有丰富且复杂的纳米受限行为. 芳香型聚酰亚胺作为刚性聚合物的典型代表, 在电子封装领域的应用占据主导地位[33]. 借助于宽频介电谱(BDS)与动态力学分析(DMA)等常规动力学表征技术, 其本体多尺度松弛动力学行为已基本明确, 包括: 链段协同运动相关的α松弛[34-35]、苯环的振动或旋转相关的β松弛[34-36]、涉及极性基团(如C=O)局部振动的γ松弛[35-36]. 但受制于超薄膜纳克级的样品量带来的反馈信号弱、测量难等问题, 上述方法难以应用于PI纳米超薄膜的测量.
2 结果与讨论
2.1 聚酰亚胺膜的合成与结构表征
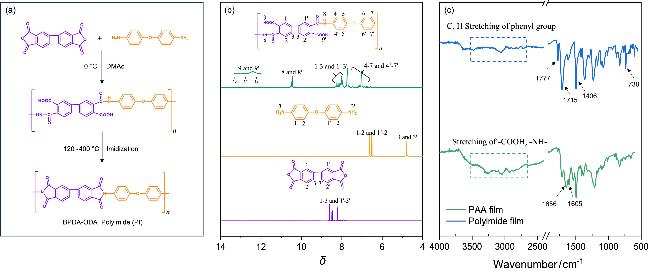
通过核磁共振氢谱(1H NMR)可以确认聚酰胺酸(PAA)的成功合成[43-44]. 如图1b所示, 谱图中δ 12~14处的宽峰归属于羧酸基团(-COOH)的质子信号, δ 10.5附近的特征峰来自于酰胺键(-C(O)NH)中的质子共振. 此外, δ 7.8~8.5与6.5~7.8范围内的信号峰分别来源于BPDA和ODA的芳香环氢原子. 通过对热处理前后PAA薄膜的红外光谱进行对比, 可以确认PAA薄膜经过高温亚胺化已充分转变为PI. 如图1c所示, 未亚胺化的PAA薄膜在1656和1605 cm−1处显示出酰胺基团的典型吸收峰, 经高温亚胺化后, 上述位置的峰消失, 取而代之的是1777 cm−1(不对称C=O伸缩振动)和1715 cm−1(对称C=O伸缩振动)处的酰亚胺环羰基吸收峰. 同时, 730 cm−1(酰亚胺C—N的弯曲振动)及1406 cm−1(酰亚胺环中C—N伸缩振动)处吸收峰的出现, 以及2500~3500 cm−1处羧基和亚氨基吸收峰的完全消失[45], 表明PAA已完全转化为聚酰亚胺. 上述结果表明我们成功制备了不同厚度的平整的PI薄膜样品.
2.2 聚酰亚胺的本体松弛
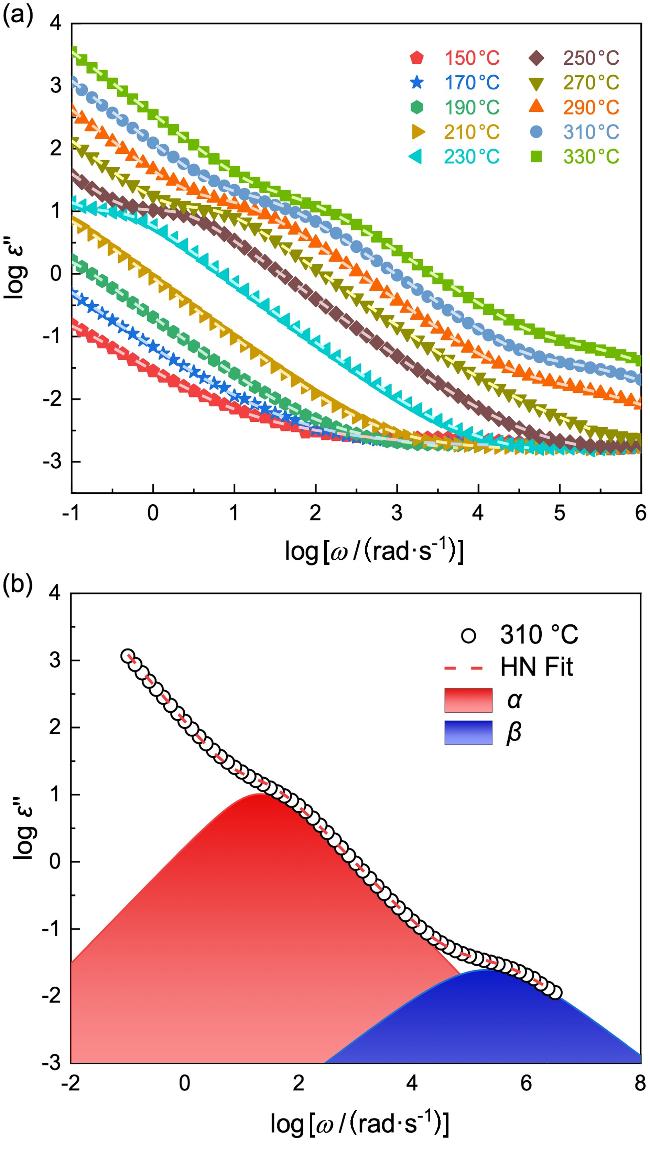
对微米级厚度PI薄膜进行介电松弛和动态热机械表征, 进而研究本体PI的松弛行为. 图2a展示的是80 μm PI膜在不同温度下(150~330 ℃)的宽频介电谱(Broadband dielectric spectroscopy, BDS). ε"代表介电损耗、ω为角频率. 可以看到, 在相同温度下, ε"随ω的减小而增大. ε"(ω)包含了PI分子多级松弛的贡献, 包括链段运动主导的α松弛和局部基团运动的次级松弛等. 此外, 随着温度的升高, ε"(ω)向高频移动, 说明PI膜的松弛速率随温度升高而加快.
$\varepsilon _{\text{NH}}^{*}(\omega ){{\varepsilon }_{\infty }}\sum\limits_{j=\alpha,\beta,\cdots }{\frac{\Delta {{\varepsilon }_{j}}}{{{[1{{(i\omega {{\tau }_{j}})}^{{{m}_{j}}}}]}^{{{n}_{j}}}}}}\frac{{{\sigma }_{ac}}}{i{{\varepsilon }_{0}}\omega }$
其中ε0=8.85×10−12 F/m为真空介电常数; ε∞和σac分别是高频介电常数和低频下的交流电导; 中间各项分别对应各松弛模式(标记为j, 对应着α和β等松弛模式)的介电强度(Δεj)和特征松弛时间(τj), 及其它们各自的对称(mj)和不对称增宽参数(nj). εHN"(ω)为εHN*(ω)的虚部. 图2为利用公式1对310 ℃下ε"(ω)的拟合结果. 拟合结果分离出了α和β松弛两种模式, 它们的特征松弛时间分别为τα=0.047 s和τβ=4.8×10−6 s; 同时, 两个松弛峰都仅表现出对称增宽, mα=0.87, mβ=0.65, nα=nβ=1.
${{\tau }_{\alpha }}{{\tau }_{\alpha 0}}\exp \frac{D{{T}_{V}}}{T{{T}_{V}}}$
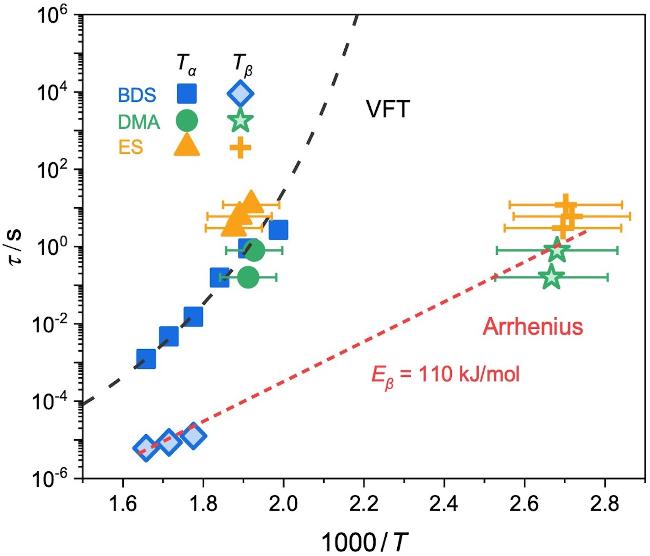
图3 PI膜的α和β松弛时间与1000/T的关系图. 黑色虚线为VFT方程拟合的结果(公式2), 红色虚线为Arrhenius方程拟合的结果(公式3)Figure 3 α and β relaxation time plotted as a function of the inverse temperature for polyimide film. The dotted lines (red and black) are the fitting of the data to the VFT and Arrhenius equation, respectively |
${{\tau }_{\beta }}{{\tau }_{\beta 0}}\exp \frac{{{E}_{\beta }}}{RT}$
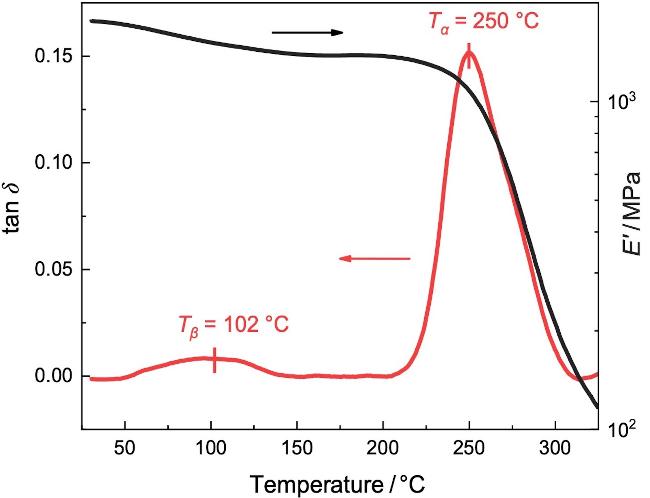
动态力学分析(Dynamic mechanical analysis, DMA)能测定PI膜在正弦交变载荷下的力学响应和损耗, 从而表征高分子松弛行为[34,52]. 图4为通过升温过程中上述80 μm PI膜的储能模量(E')和损耗正切角(tan δ)的变化. 如图所示, PI膜的E'随温度升高出现了两次台阶式下降: 分别在50~150 ℃和250~300 ℃处; 同时, 对应着两个tan δ的损耗峰. 它们可以分别归属于升温过程中PI薄膜内的基团局部运动对应的β松弛模式和链段运动所贡献的α松弛模式. 特征松弛温度(Tα和Tβ)可由tan δ峰值温度来确定, 即Tβ=102 ℃和Tα=250 ℃. 此外, 损耗峰峰值处, 材料的松弛时间等于实验测量时间; DMA的实验时间由其频率决定, 即: τDMA=1/2πfDMA. 那么, 可以将DMA确定的α松弛(Tα,DMA, τDMA)和β松弛(Tβ,DMA, τDMA)汇总到图3中(见图中的绿色符号). 可见, DMA的结果与BDS结果很好吻合, 也验证了BDS和DMA二者测量结果的准确性.
另一方面, PI薄膜受热会发生膨胀, 导致膜厚随温度升高而增大. 热膨胀能力用热膨胀系数(k)表示. 温度升高将加速分子运动; 当薄膜内某一特定运动模式被激活, 材料的k也会随之增大. 利用变温椭圆偏振光谱(Ellipsometry, ES)对300 nm厚度PI膜的厚度(h)随温度的变化进行了研究, 结果见图5. PI薄膜表现出典型的线性膨胀行为; 薄膜厚度随温度升高而线性增厚. 为了定量描述PI薄膜的热转变, 对h~T曲线求导得到薄膜k, 即
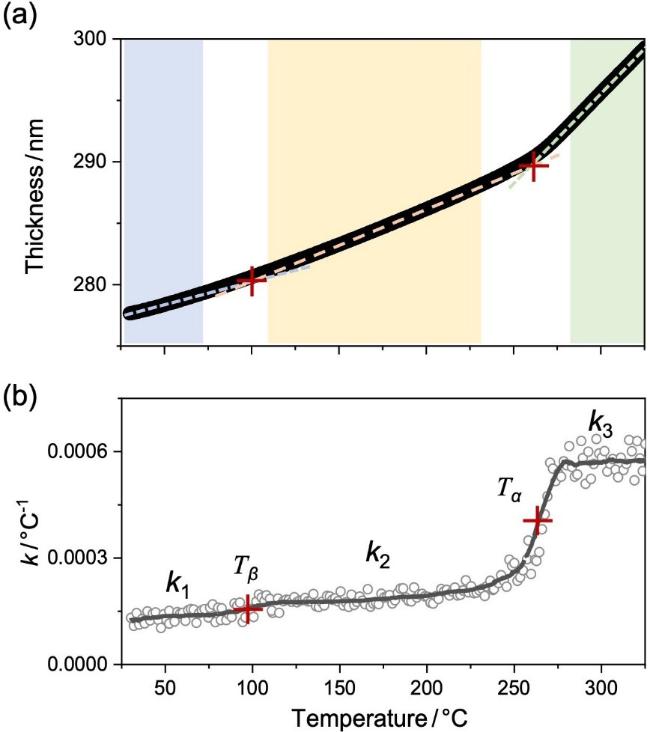
图5b显示了PI薄膜的k随温度上升的变化行为. 可以看到, 当T<70, 120<T<240和T>270 ℃范围内, k维持不变, 分别为k1≈0.00013、k2≈0.00019、k3≈0.00058 ℃−1. 而在100和260 ℃附近, k增大, 对应着两个松弛转变. 在100 ℃附近时, PI薄膜的k增幅较小, k2≈1.5k1, 说明此温度下所激活的分子运动能力较弱, 对应局域小尺度运动的β松弛, 即Tβ,ES≈99 ℃; 而在260 ℃附近时, k3≈3k2, 表明有较大尺度的运动模式被激活, 对应着链段运动的α松弛. Tβ,ES≈99 ℃和Tα,ES≈260 ℃与DMA测得α和β松弛温度一致. 同时, 变温椭圆偏振光谱的实验时间尺度由线性升温速率(q)决定, 即τES=1/q. 那么, 可将变温椭圆偏振光谱所确定的α松弛(Tα,ES, τES)和β松弛(Tβ,ES, τES)汇总到图3中(见图中的绿色符号). 由图3可见, 椭圆偏振光谱(ES)的测量结果与常规方法BDS和DMA的测量结果基本一致. 这说明变温椭圆偏振光谱能够准确测定本体PI膜的多尺度松弛动力学.
2.3 纳米聚酰亚胺超薄膜的松弛
椭圆偏振光谱对薄膜厚度的测定具有亚纳米级的超高分辨率, 故可以将此法用于研究厚度低于100 nm的超薄膜松弛行为, 从而弥补DMA或BDS等常规动力学方法难以测量纳米薄膜的局限.
图6a为变温椭圆偏振光谱测量的不同厚度PI薄膜的热膨胀行为. 当薄膜厚度大于100 nm时, 聚酰亚胺膜表现出相同的热膨胀行为; 100 nm和300 nm聚酰亚胺膜的h/h0~T完全重合. 当膜厚降低到74 nm左右时, 薄膜热膨胀行为开始偏离厚膜: 随着膜厚降低, 薄膜的热膨胀变得更显著. 对于6 nm厚度的PI超薄膜, 可以观察到h/h0~T曲线斜率发生了两次转变, 对应着α和β两个松弛转变. 这也说明椭圆偏振光谱是研究超薄膜动态松弛过程的有效方法.
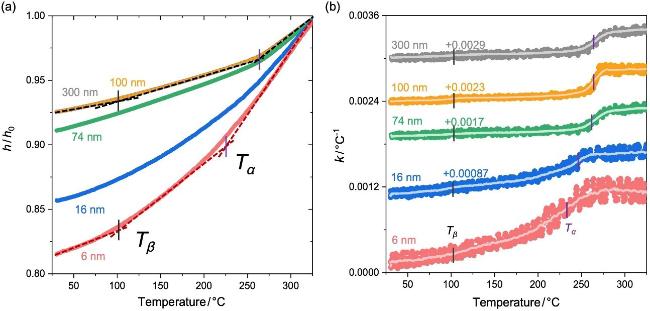
图6 PI薄膜的(a)归一化厚度(h/h0, h0为薄膜在330 ℃下的膜厚)和(b)膨胀系数随温度的变化. 图(b)中的曲线经过纵向平移, 以更好地展示其差异Figure 6 (a) Normalized thickness (h/h0, h0 is the thickness of films at 330 ℃) and (b) thermal expansion coefficient as a function of temperature for polyimide ultrathin films. The curves in panel (b) have been vertically shifted for better presentation of their difference |
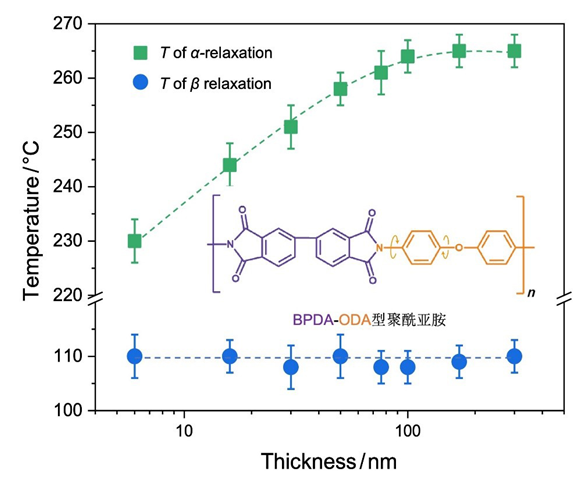
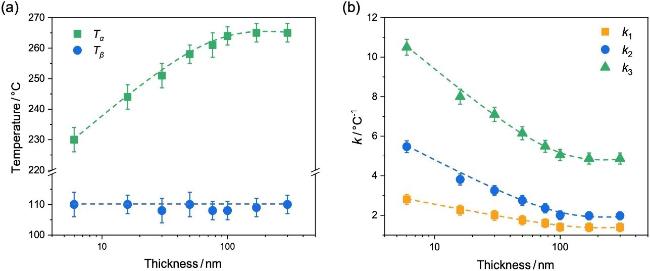
图6b为不同厚度PI薄膜的k与温度的依赖性. 随着PI薄膜的膜厚降低, Tα明显降低, 转变的温度区域增宽; 而β松弛温度Tβ几乎不发生变化. 图7a总结了PI薄膜Tβ与Tα与薄膜厚度的关系. 结果表明, 当膜厚大于约100 nm时, 薄膜的Tα不随膜厚而变化; 继续降低膜厚, Tα随着薄膜厚度的降低而减小. 这表明纳米受限加速了PI薄膜的链段松弛. 然而, PI薄膜的Tβ却不随膜厚的变化而改变. 薄膜Tα的纳米受限效应来源于自由表面的作用. 自由表面附近的链段缺少周围分子链段的相互作用, 协同性与耦合作用减弱, 运动加快; 随着膜厚降低, 快的表面链段贡献增加从而加速薄膜的松弛, 使Tα降低[53-55]. 而β松弛仅涉及局部的基团运动, 不参与链段间的协同与耦合[55-56]; 因此, β松弛不受自由表面所带来的链段运动耦合性改变的影响, Tβ不表现出膜厚依赖性[57].
3 结论与展望
本工作制备了从微米级到纳米级的芳香型BPDA-ODA聚酰亚胺薄膜, 利用宽频介电谱、动态力学分析以及变温椭圆偏振光谱研究了PI本体和纳米薄膜的多级松弛行为. 结果表明PI在270和100 ℃附近分别出现链段运动的α松弛和芳香环转动的β松弛过程. 并且, 随着PI薄膜厚度降低至100 nm, PI的α转变温度 (Tα)降低, 而β转变温度(Tβ)基本不变, 热膨胀系数显著增大. 这一结果说明PI薄膜的多级松弛行为存在不同的膜厚依赖性. 该结果对于PI薄膜的应用具有重要意义. 一方面, PI超薄膜升高的膨胀系数降低了薄膜热稳定性, 可能在薄膜与器件界面产生热应力, 造成薄膜剥离甚至引起器件翘曲和断裂. 另一方面, 纳米PI薄膜降低的Tg可能加速薄膜在使用过程中发生物理老化, 从而使得薄膜变脆、表面产生银纹等, 造成器件失效. 因此, 开发具有低热膨胀系数和高Tg的高稳定PI超薄膜对于精密器件的设计和制备至关重要. 如何调控纳米PI薄膜的松弛和热转变行为, 抑制薄膜热膨胀将是未来PI薄膜研究的重要方向.
4 实验部分
4.1 材料
联苯四甲酸二酐(biphenyltetracarboxylic diand- hydride, BPDA, AR)、4,4'-二氨基二苯醚(4,4'-oxydi- aniline, ODA, AR)、N,N-二甲基乙酰胺(N,N-dimethyl-acetamide, DMAc)、甲醇(AR)均购自上海麦克林生化科技股份有限公司, 直接使用.
4.2 聚酰胺酸(Polyamide Acid, PAA)膜的制备
利用两步法制备BPDA-ODA型聚酰亚胺薄膜(图1). 首先是聚酰胺酸(PAA)溶液的合成: 氮气保护下, 在250 mL三口玻璃中将30.0 mmol的ODA溶解于120 mL的DMAc形成均相溶液. 利用冰水浴将反应体系温度控制在0 ℃附近, 向溶液中一次性加入28.0 mmol的BPDA粉末, 搅拌至完全溶解后再加入20 mL DMAc并持续反应2~3 h. 最后, 再分批加入2.0 mmol的BPDA以调节聚合度, 获得粘稠的PAA/DMAc溶液. 利用甲醇沉淀产物, 并将其再次溶解于DMAc, 反复多次纯化上述产物以除去未反应单体, 得到淡青色的PAA固体.
利用浇铸法和旋涂法(Spin-coating)制备不同厚度聚酰胺酸(PAA)薄膜: 制备微米级PAA膜时采用浇铸成膜法, 将PAA溶液(20% (w))均匀涂覆在洁净的玻璃板上, 在洁净工作台放置24 h待溶剂自然挥发成膜; 制备纳米级PAA超薄膜时采用旋涂成膜法, 将PAA溶液涂覆于硅片表面, 以3000 r/min转速旋涂20 s, 通过调节PAA溶液浓度得到不同厚度PAA纳米超薄膜.
4.3 PAA膜高温亚胺化制备聚酰亚胺薄膜
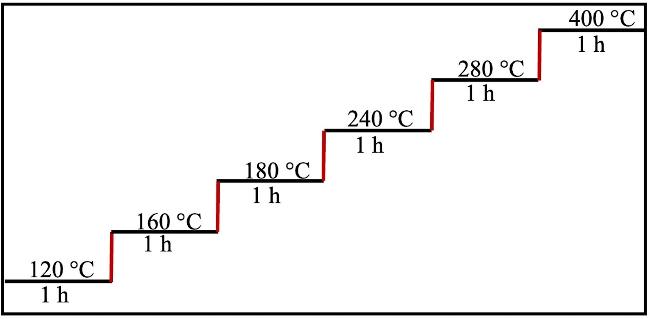
图8 PAA薄膜高温亚胺化制备PI薄膜的升温程序图Figure 8 Temperature protocol for preparing PI films by imidization of PAA films at high temperature |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.4 表征方法
核磁共振氢谱(1H NMR): 采用德国Bruker公司的AVANCEII/400型核磁共振波谱仪进行测试, 溶剂采用氘代二甲基亚砜(DMSO-d6, 溶剂峰化学位移为2.50).
全反射傅里叶变换红外光谱(ATR FT-IR): 使用美国ThermoFisher Scientific公司的Nicolet 5700型红外光谱仪进行测试, 波数扫描范围为500~4000 cm−1, 扫描次数为32次.
宽频介电谱(BDS): 采用德国Novocontrol公司Concept 50型介电谱仪进行测试, 测试温度范围为150~330 ℃, 每间隔20 ℃进行等温扫频测试, 频率范围10−1~107 Hz.
动态力学分析(DMA): 使用瑞士Mettler Toledo公司的DMA1型动态力学分析仪, 以速率3 ℃/min在30~330 ℃温度范围内进行线性升温测试. PI膜裁剪成40 mm×5 mm×80 μm的片状样条. 测试采用拉伸模式, 样条的有效拉伸长度为30 mm; 控制测试的正弦拉伸幅度为10 μm (应变振幅为0.03%), 正弦频率分别为fDMA=0.2, 1及3 Hz.
变温椭圆偏振光谱: 采用美国J. A. Woollam公司的M-2000V型椭圆偏振光谱仪进行测试, 波长(λ)覆盖350~1500 nm. 实验中, 使用英国Linkam公司的热台对薄膜样片进行控温, 分别以q=5, 10及20 ℃/min的速率在30~330 ℃的范围进行线性升温. 同时, 利用椭圆偏振光谱仪原位测定薄膜的光谱Ψ(λ)和Δ(λ); 其中Ψ(λ)和Δ(λ)分别是经薄膜反射后, 两个偏振分量的入射光线和反射光线之间的振幅改变和相位改变. 椭圆偏振光谱Ψ(λ)和Δ(λ)经过光学模型的拟合处理可以给出薄膜的厚度(h), 其厚度分辨率精确到0.1 nm.
原子力显微镜(AFM): 使用美国Bruker公司的ICON Fastscan型原子力显微镜. 测试采用轻敲模式(tapping mode); 扫描结果使用NanoScope Analysis软件进行处理和分析.
(Cheng, B.)