

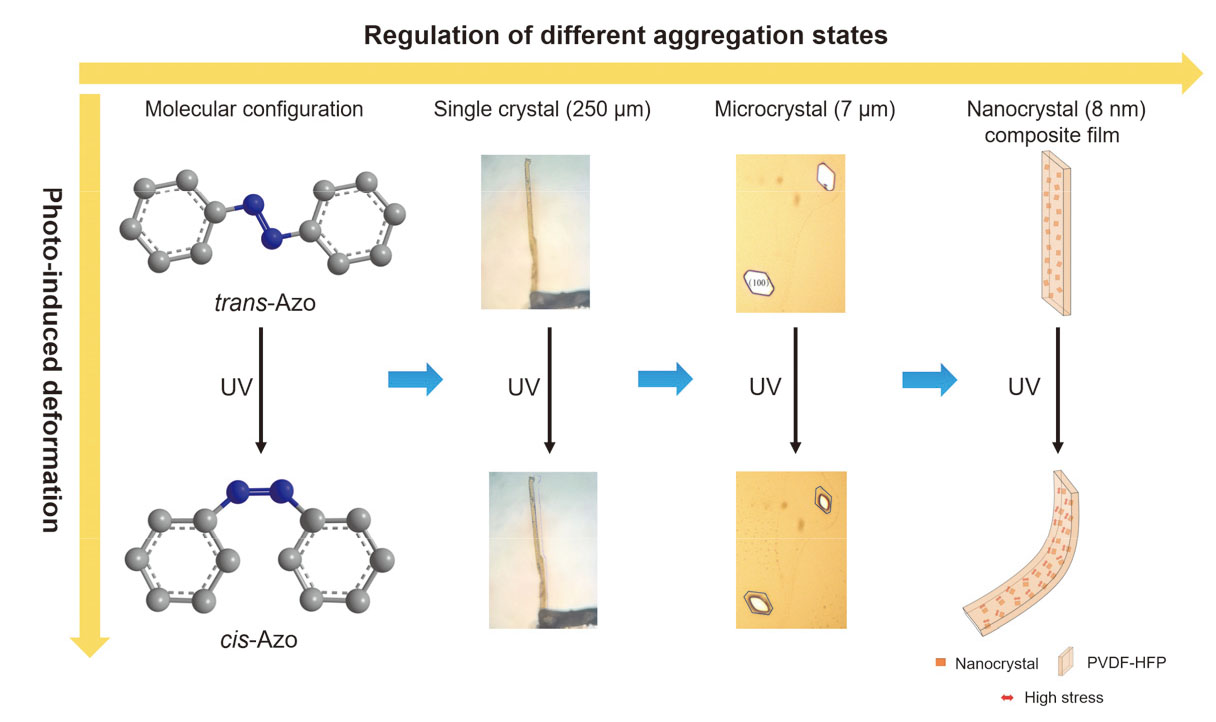
1 引言
2 结果与讨论
2.1 Azo光致异构化原理探究
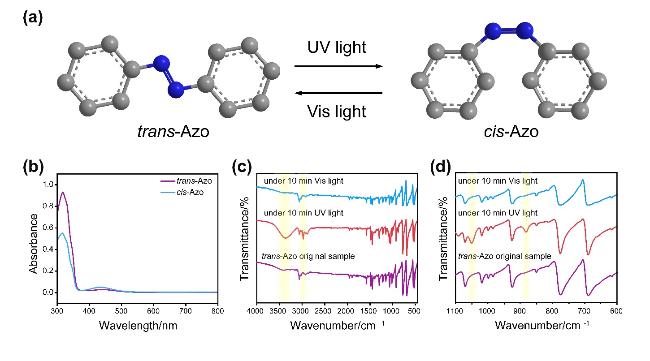
2.2 Azo晶体光响应行为分析
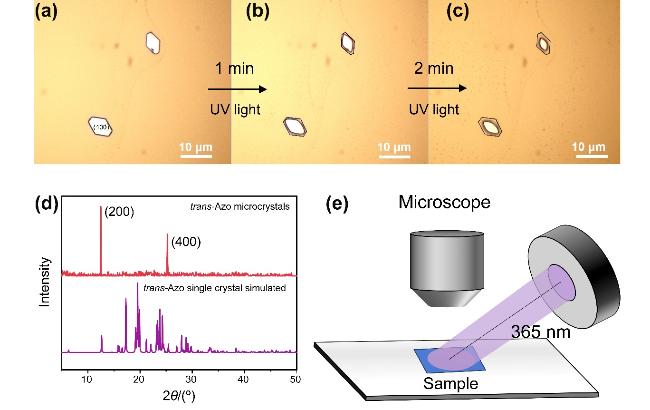
图2 (a~c) Azo微晶在紫外光照射1、2 min前后的光学显微镜形貌图; (d) trans-Azo 微晶与单晶模拟的XRD对比图谱; (e) Azo微晶光致响应行为探究实验设置示意图Figure 2 (a~c) Optical microscopy image of Azo microcrystals before and after UV light exposure for 1 and 2 min; (d) XRD comparison diagram between trans-Azo microcrystals and trans-Azo single crystal simulated; (e) Schematic diagram of experimental set-up for investigating photoresponse behavior of Azo microcrystals |
2.3 Azo晶体复合薄膜光致动原理及其执行行为探究

图3 (a) Azo/PVDF-HFP-drop casting薄膜的TEM图; (b) Azo/PVDF-HFP-drop casting薄膜的HR-TEM图, 插图内为SAED图; (c~e) PVDF-HFP, Azo/PVDF-HFP-spin coating, Azo/PVDF-HFP-drop casting薄膜的XRD, UV-vis图谱以及弯曲角度随时间变化曲线图; (f) Azo/PVDF-HFP-drop casting薄膜的循环弯曲曲线图Figure 3 (a) TEM image of Azo/PVDF-HFP-drop casting film; (b) HR-TEM image of Azo/PVDF-HFP-drop casting film, with the inset showing SAED image; (c~e) XRD diagram, UV-vis spectra and bending angle curves change with time; (f) Cyclic bending curve of Azo/PVDF-HFP-drop casting film |
 ), (
), (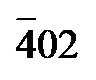 )和(400)晶面的衍射峰, 而其他两种则无. 这表明只有Azo/PVDF-HFP-drop casting成功保留了Azo纳晶良好的结晶性, 而Azo/PVDF-HFP- spin coating为无定形态. 这种差异主要源于两种制备工艺的本质区别. 滴铸法是一种缓慢且静态的工艺, 能够促使纳米晶在聚合物中聚集, 从而形成具有高结晶度的薄膜. 相比之下, 旋涂法则是一种非平衡态的工艺. 在旋涂过程中, 溶液滴加在高速旋转的基底上迅速甩出并蒸发, 导致分子缺乏足够的时间进行有序排列, 从而有效抑制了结晶过程的发生, 最终导致旋涂薄膜的结晶度通常相对较低[44]. 随后对三种薄膜进行UV-vis吸收测试, 测试结果如图3d所示, Azo/PVDF-HFP-drop casting与Azo/PVDF-HFP-spin coating均表现出与Azo分子类似的吸收特征曲线, 对360 nm后的紫外光有着良好的吸收能力. 而对于PVDF-HFP薄膜, 虽然也在紫外光区具有一定的吸收, 但吸收强度显著弱于前两种复合薄膜, 不足以对复合薄膜的响应能力造成较大影响. 随后, 在波长为365 nm紫外灯的照射下对三种薄膜的光致驱动能力进行了测试. 测试结果如图3e所示, PVDF-HFP薄膜未展现出响应能力, Azo/PVDF-HFP-spin coating复合薄膜虽能响应紫外光, 但是致动效果弱且不稳定, 而Azo/PVDF-HFP-drop casting可以在紫外光照射后的20 s内以先快后慢的速度弯曲到21.13°, 展现出了稳定、高效的致动能力. 此外, 纳晶复合薄膜可以随着Azo晶体颗粒的可逆异构化而循环形变. 具体如图3f所示, Azo/PVDF-HFP-drop casting在紫外灯的照射下可以在15~20 s内达到15°~20°的弯曲形变. 当关闭紫外灯后, 由于Azo纳晶颗粒在可见光作用下由cis构象缓慢恢复到trans构象, 纳晶复合薄膜也在1.5~2.5 min内缓慢恢复到初始状态. Azo/PVDF-HFP-drop casting的可逆致动至少能够维持5次循环, 并随着循环次数的增多, 最大弯曲角度逐渐增大, 响应时间逐渐缩短.
)和(400)晶面的衍射峰, 而其他两种则无. 这表明只有Azo/PVDF-HFP-drop casting成功保留了Azo纳晶良好的结晶性, 而Azo/PVDF-HFP- spin coating为无定形态. 这种差异主要源于两种制备工艺的本质区别. 滴铸法是一种缓慢且静态的工艺, 能够促使纳米晶在聚合物中聚集, 从而形成具有高结晶度的薄膜. 相比之下, 旋涂法则是一种非平衡态的工艺. 在旋涂过程中, 溶液滴加在高速旋转的基底上迅速甩出并蒸发, 导致分子缺乏足够的时间进行有序排列, 从而有效抑制了结晶过程的发生, 最终导致旋涂薄膜的结晶度通常相对较低[44]. 随后对三种薄膜进行UV-vis吸收测试, 测试结果如图3d所示, Azo/PVDF-HFP-drop casting与Azo/PVDF-HFP-spin coating均表现出与Azo分子类似的吸收特征曲线, 对360 nm后的紫外光有着良好的吸收能力. 而对于PVDF-HFP薄膜, 虽然也在紫外光区具有一定的吸收, 但吸收强度显著弱于前两种复合薄膜, 不足以对复合薄膜的响应能力造成较大影响. 随后, 在波长为365 nm紫外灯的照射下对三种薄膜的光致驱动能力进行了测试. 测试结果如图3e所示, PVDF-HFP薄膜未展现出响应能力, Azo/PVDF-HFP-spin coating复合薄膜虽能响应紫外光, 但是致动效果弱且不稳定, 而Azo/PVDF-HFP-drop casting可以在紫外光照射后的20 s内以先快后慢的速度弯曲到21.13°, 展现出了稳定、高效的致动能力. 此外, 纳晶复合薄膜可以随着Azo晶体颗粒的可逆异构化而循环形变. 具体如图3f所示, Azo/PVDF-HFP-drop casting在紫外灯的照射下可以在15~20 s内达到15°~20°的弯曲形变. 当关闭紫外灯后, 由于Azo纳晶颗粒在可见光作用下由cis构象缓慢恢复到trans构象, 纳晶复合薄膜也在1.5~2.5 min内缓慢恢复到初始状态. Azo/PVDF-HFP-drop casting的可逆致动至少能够维持5次循环, 并随着循环次数的增多, 最大弯曲角度逐渐增大, 响应时间逐渐缩短.2.4 不同Azo质量分数的Azo/PVDF-HFP纳晶复合薄膜表征及执行效果研究
), (), (400)晶面的衍射峰, 且随着Azo质量分数的提高, Azo纳晶颗粒数量逐渐增多, 结晶峰强度增强. 进一步通过拉伸仪对不同Azo质量分数的纳晶复合薄膜进行了力学性能的测试. 如支持信息图S12所示, 随着Azo质量分数的提高, 薄膜逐渐变脆, 而在Azo占比达到65%时延展性突然增强, 这可能与孔隙对薄膜表面形貌带来的显著破坏有关.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图4 (a, b)不同Azo质量分数的Azo/PVDF-HFP-drop casting薄膜弯曲角度随时间变化曲线图与最大弯曲角度统计图; (c)质量分数为40%的Azo/PVDF-HFP-drop casting薄膜的执行过程Figure 4 (a, b) Bending angle curves change with time and maximum bending angle statistical diagram of Azo/PVDF-HFP-drop casting films with different Azo mass ratios; (c) Actuation process of Azo/PVDF-HFP-drop casting film with 40% mass ratio of Azo content |