

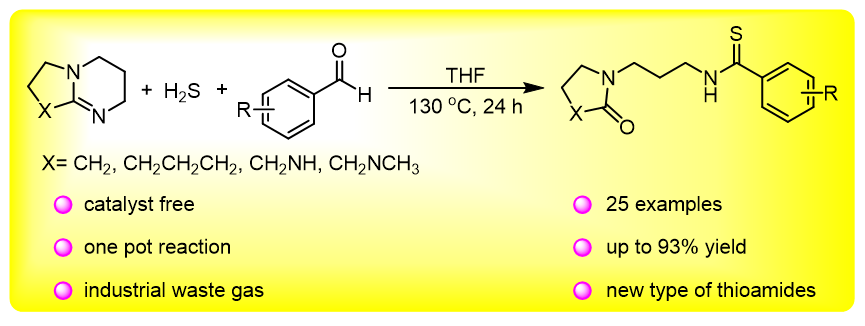
1 引言
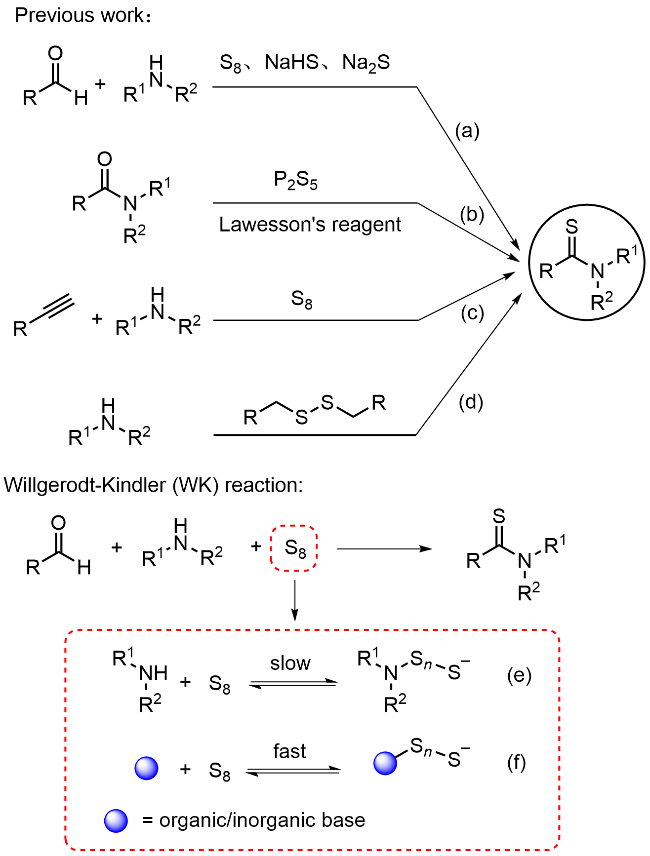
目前, 已报道了多种合成硫代酰胺类化合物的方法(Scheme 2): (a)醛、胺和硫源组成的Willgerodt-Kindler (WK)反应[18-21]; (b)酰胺与五硫化二磷或劳森试剂之间的氧硫交换反应[22-24]; (c)炔烃、胺与硫磺反应[25-26]; (d)二硫化物与胺反应[27-28]等. 其中, WK反应是以醛、胺、硫磺为原料合成硫代酰胺的经典方法. 该反应体系通常以硫磺(S8)作为硫源, 尽管S8可通过开环形成多硫阴离子作为活性亲核试剂, 但其自发解离速率较慢[29-31] (Scheme 2e). 为加速多硫阴离子的生成, 通常会在反应中加入碱性助剂或催化剂以活化S8形成多硫阴离子[32-36](Scheme 2f). 因此, 如何高效合成硫代酰胺成为众多科研工作者的重要研究课题[32,37-39].
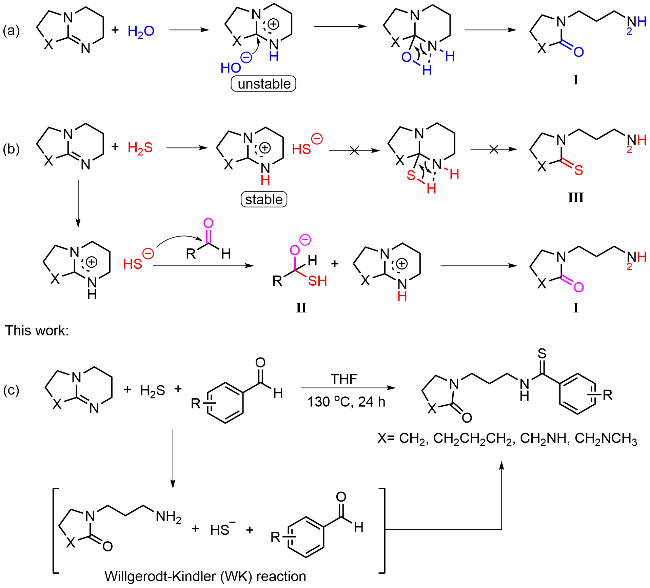
在硫代酰胺的合成中, 由于硫磺、硫化钠和硫氢化钠等传统硫源在有机溶剂中的溶解性能差而受到限制, 而劳森试剂则面临原子利用率低的瓶颈. 这些局限性严重制约了其大规模应用. 与此同时, 随着工业化的快速推进, 硫化氢(H2S)作为化石燃料加工过程中的副产物产量持续增加, 但目前主要通过克劳斯工艺转化为硫磺, 经济效益有限[40-42]. 针对这一现状, 开发以H2S为硫源合成高附加值含硫化合物的新方法具有重要意义[43-44]. 从酸碱中和的角度来看, H2S作为一种酸性气体, 只需引入碱性溶剂或溶液即可被快速吸收. 据文献报道, 环脒(CA)类碱性化合物可快速吸收H2S生成稳定的CAH⁺HS⁻[45-46]. 值得注意的是, 环脒类化合物在含水条件下可以开环形成胺[47-52](Scheme 3a), 该过程首先由环脒与水形成不稳定的CAH⁺OH⁻, 随后O⁻离子亲核进攻CAH⁺完成开环形成胺Ⅰ. 然而CAH⁺HS⁻较稳定, 无法通过HS⁻离子亲核进攻CAH⁺形成胺Ⅲ (Scheme 3b). 鉴于HS⁻作为亲核性硫源可以亲核进攻醛羰基形成含有O⁻离子的中间体Ⅱ, 我们设想该中间体Ⅱ可以通过O⁻离子亲核进攻CAH⁺使其开环形成胺Ⅰ, 随后与醛、HS⁻发生三组分WK反应, 进而开发硫代酰胺类衍生物的高效合成方法(Scheme 3c). 在该反应体系中, H2S提供的H⁺促进环脒开环生成氨基源, 同时释放的HS⁻作为硫源参与反应, 而开环后生成的胺又能维持反应体系的碱性环境, 最终成功制备了目标产物硫代酰胺. 这一策略不仅实现了H2S的高值化利用, 还为硫代酰胺的高效合成提供了新思路.
2 结果与讨论
2.1 反应条件的优化
本研究以DBU (1a)和苯甲醛(2a)为模型底物, H2S为硫源, 系统考察了投料比、温度、溶剂和时间对反应的影响规律(Table 1). 首先对投料比进行了优化(Table 1, Entries 1~8), 实验结果表明, 当DBU、苯甲醛和H2S的投料物质的量比为1∶2∶4时, 反应产率最高达83% (Table 1, Entry 4). 随后考察了温度对反应的影响规律(Table 1, Entries 4, 9~14), 随着反应温度升高, 产率呈现先增加后减小的趋势. 当反应温度升高至130 ℃时, 反应产率高达83% (Table 1, Entry 4). 由于该反应涉及气液两相体系, 溶剂的选择至关重要. 实验发现, 四氢呋喃(Table 1, Entry 4)、1,4-二氧六环(Table 1, Entry 15)等醚类溶剂表现出良好的反应效果. 相比之下, 聚乙二醇400 (Table 1, Entry 16)作为溶剂时, 由于其高粘度不利于反应进行, 反应产率只有20%. 当使用N,N-二甲基甲酰胺(DMF)作为溶剂时(Table 1, Entry 17), 产率降低至8%, 且在分离过程中检测到N,N-二甲基硫代苯甲酰胺, 推测原因可能是DMF在高温下分解生成二甲胺, 二甲胺会与苯甲醛、H2S反应生成N,N-二甲基硫代苯甲酰胺, 使反应产率降低. 当溶剂为N-甲基吡咯烷酮(NMP)时(Table 1, Entry 18), 产率只有35%. 由于二甲基亚砜(DMSO)本身具有氧化性, 其作为溶剂可能将苯甲醛氧化为苯甲酸[53], 导致没有产物生成(Table 1, Entry 19). 当溶剂为乙腈时(Table 1, Entry 20), 其腈基易受HS⁻亲核进攻, 不利于反应进行. 综合比较, 最终选定THF作为最佳反应溶剂. 最后, 考察了反应时间对产率的影响规律(Table 1 Entry 4, 21~23). 结果表明, 随着反应时间延长, 产率呈现先增加后趋于平稳的趋势, 当反应时间达到24 h时, 产率稳定在83%.
表1 反应条件筛选Table 1 Optimization of the reaction |
| Entry | Feed ratioa | Temp./℃ | Time/h | Yieldb/% |
|---|---|---|---|---|
| 1 | 1∶2∶1 | 130 | 24 | trace |
| 2 | 1∶2∶2 | 130 | 24 | 7 |
| 3 | 1∶2∶3 | 130 | 24 | 56 |
| 4 | 1∶2∶4 | 130 | 24 | 83 |
| 5 | 1∶2∶5 | 130 | 24 | 70 |
| 6 | 1∶1∶4 | 130 | 24 | 51 |
| 7 | 1∶3∶4 | 130 | 24 | 71 |
| 8 | 1∶4∶4 | 130 | 24 | 72 |
| 9 | 1∶2∶4 | 80 | 24 | 9 |
| 10 | 1∶2∶4 | 90 | 24 | 27 |
| 11 | 1∶2∶4 | 100 | 24 | 53 |
| 12 | 1∶2∶4 | 110 | 24 | 57 |
| 13 | 1∶2∶4 | 120 | 24 | 74 |
| 14 | 1∶2∶4 | 140 | 24 | 80 |
| 15c | 1∶2∶4 | 130 | 24 | 74 |
| 16d | 1∶2∶4 | 130 | 24 | 20 |
| 17e | 1∶2∶4 | 130 | 24 | 8 |
| 18f | 1∶2∶4 | 130 | 24 | 35 |
| 19g | 1∶2∶4 | 130 | 24 | trace |
| 20h | 1∶2∶4 | 130 | 24 | trace |
| 21 | 1∶2∶4 | 130 | 12 | 64 |
| 22 | 1∶2∶4 | 130 | 36 | 79 |
| 23 | 1∶2∶4 | 130 | 48 | 84 |
Reaction conditions: DBU (1.0 mmol.), Solvent: THF (2 mL). a Feed ratio (mmol)=DBU∶Benzaldehyde∶H2S; b Isolated yield; c1,4-Dioxane (2 mL); d PEG400 (2 mL); e DMF (2 mL); f NMP (2 mL); g DMSO (2 mL); h CH3CN (2 mL). |
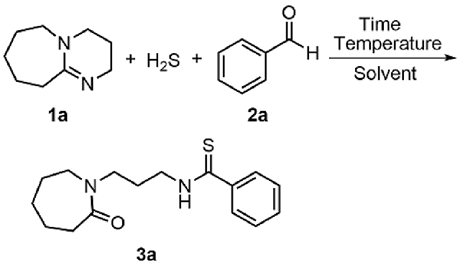
2.2 底物适用范围考察
在最优反应条件下, 本工作系统考察了不同醛类和H2S、环脒反应合成硫代苯甲酰胺方法的适用性(Scheme 4). 结果表明, 该方法对反应底物适应范围较广, 但是不同取代基的反应性也有差别. 首先以DBU作为底物, 考察了醛类衍生物的适用范围. 结果发现, 当苯甲醛连有给电子基团时(3b~3g), 反应产率较高. 但是羟基为取代基时, 其产率较低(3e, 3f), 其原因可能是苯酚具有酸性, 本身可以提供质子, 抑制了反应进行. 当苯甲醛连有吸电子基团(3h~3k), 产率较低. 推测是由于吸电子基使苯环上电子云密度减小, 可以增强醛基上羰基碳的正电性. 虽然能够促进HS⁻亲核进攻苯甲醛生成中间体Ⅱ (Scheme 3b), 但由于O⁻的亲核性也会降低, 最终会使反应效果变差, 导致产率降低. 值得注意的是, 当4位为腈基时, 腈基也可以与硫化氢反应得到另一种硫代酰胺(3j'). 在考察杂环醛类化合物时(3l~3q), 发现除2-吡啶甲醛能以优异的产率获得目标产物(3l)外, 其余杂环醛的产率相对较低. 随后, 研究进一步考察了1,5-二氮杂双环[4.3.0]九碳-5-烯(DBN)和1,5,7-三氮杂双环 [4.4.0] 癸烯-5-烯(TBD)的反应性能. 实验结果表明, DBN和TBD同样可与醛、H2S反应生成目标产物, 但其反应效果较DBU略差, 仅能以中等产率获得目标化合物(3r~3x). 然而, 当以7-甲基-1,5,7-三氮杂二环[4.4.0]癸-5-烯(MTBD)为原料时, 仅能以8%的产率获得目标产物(3y), 推测其可能由于7-甲基的空间位阻使其反应性降低.
2.3 控制实验
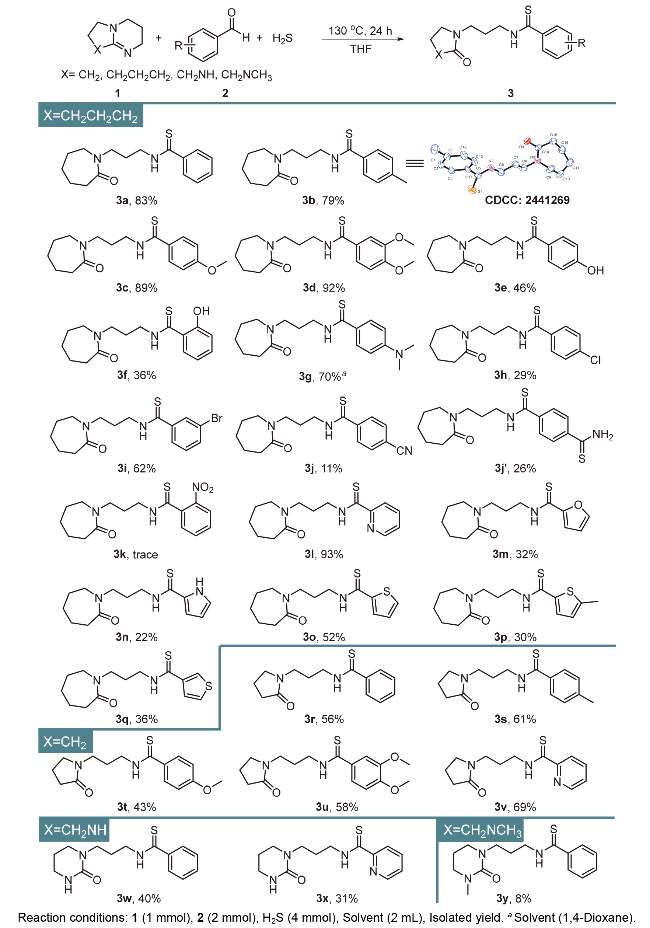
根据文献报道, DBU在含水环境中可能发生水解开环反应[52]. 为排除溶剂中水分对该反应的影响, 本研究在无溶剂条件下(Scheme 5B), 通过DBU、苯甲醛和H2S的一锅法反应, 以45%的产率获得目标产物. 与以水为溶剂的反应体系(Scheme 5C)相比, 两者产率相当, 这表明水对反应进程无明显影响. 此外, 为明确产物中O原子的来源, 在N2保护且无溶剂的条件下(Scheme 5D)进行反应, 最终仍以48%的产率获得目标产物, 由于该体系仅存在DBU、苯甲醛、H2S和N2四种物质, 因此可以确定目标产物己内酰胺中的O原子必定来源于苯甲醛. 通过该对照实验, 不仅排除了水和氧气对反应的潜在影响, 同时也明确了苯甲醛作为氧原子来源的关键作用. 随后, 当以DBUH⁺HS⁻替代DBU和H2S, 在标准反应条件下与苯甲醛反应时, 仅以32%的产率获得目标产物3a (Scheme 5E). 推测产率低是由于DBUH⁺HS⁻在高温下部分解离成DBU和H2S, 使体系中缺乏足够的H⁺和硫源, 随后在该体系中加入3 mmol H2S, 最终目标化合物3a的产率提升至78% (Scheme 5F). 这一结果证明反应的起始步骤由DBU与H2S反应形成DBUH⁺HS⁻中间体. 最后, 当使用1-(3-氨基丙基)氮杂环庚烷-2-酮替代DBU作为底物, 在标准条件下与苯甲醛和H2S反应, 同样以63%的产率获得了目标产物 3a (Scheme 5G), 进一步证明该分子是参与最终成环的有效反应前体.
2.4 原位红外捕捉反应中间体
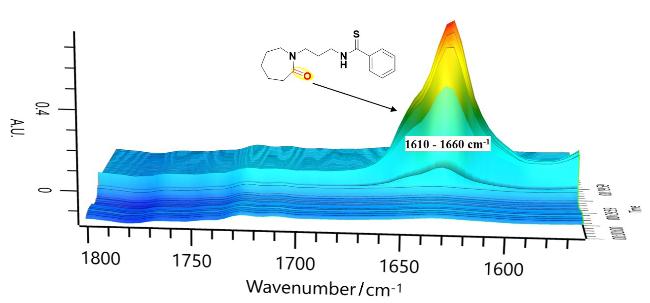
为深入探究反应机理, 本研究采用原位红外对反应中间体进行了实时监测. 如图1所示, 在反应釜中依次加入DBU和苯甲醛, 在1619 cm⁻1处观察到DBU的 C=N伸缩振动峰, 此外, 在1704 cm⁻1处为醛基C=O伸缩振动峰. 随后, 向两者的混合溶液中通入H2S气体, 结果显示通入H2S后DBU的C=N伸缩振动峰迅速消失, 推测DBU与H2S反应生成了DBUH⁺HS⁻中间体. 同时, 在1704 cm⁻1处的醛基C=O伸缩振动峰也在通入H2S后迅速消失, 由此推测HS⁻与苯甲醛发生亲核反应, 使C=O键断裂. 进一步分析发现, 在917 cm⁻1处(图2)出现了C—S键的特征信号峰, 表明C—S键的形成. 此外, 在1249 cm⁻1处观察到C—O—C不对称伸缩振动峰(图3), 推测该峰来源于苯甲醛中间体与DBUH⁺进一步反应生成的中间体. 值得注意的是, C—O—C键的信号峰在刚通入H2S时强度较弱, 但随着反应温度升高, 其强度逐渐增强, 表明温度对该中间体的生成具有显著影响. 随着反应的进行, 在1644 cm⁻1处(图1)出现了酰胺类C=O键的伸缩振动峰, 表明此时已形成含有酰胺结构的中间体. 为验证这一结果, 本研究对比了目标产物3a的原位红外谱图(图4), 在1610~1660 cm⁻1范围内观察到3a产物中己内酰胺C=O键的伸缩振动峰, 与反应液在1644 cm⁻1处出现的C=O键伸缩振动峰一致. 综上所述, 通过原位红外监测, 本研究揭示了反应过程中关键中间体的生成与转化, 为阐明反应机理提供了有力证据.
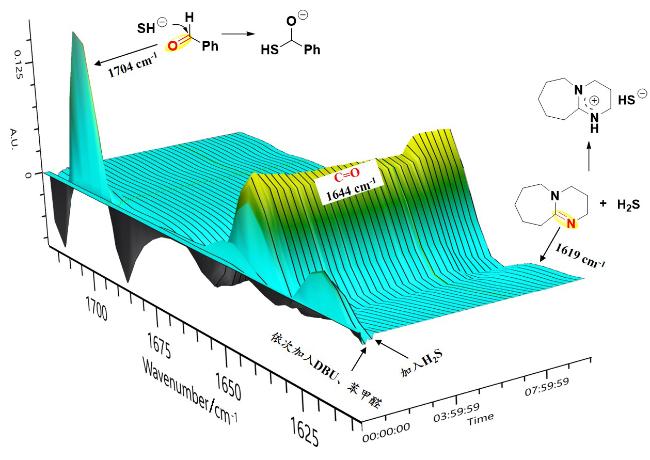
图1 反应液在1704、1644和1619 cm⁻1的原位红外光谱Figure 1 In situ FT-IR spectra of the reaction liquid at 1704, 1644 and 1619 cm⁻1 |
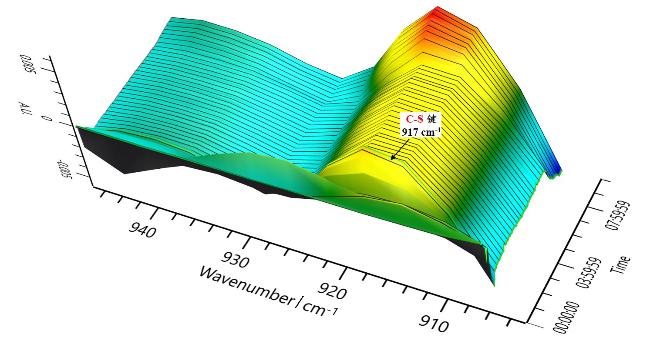
图2 反应液在917 cm⁻1的原位红外光谱Figure 2 In situ FT-IR spectra of the reaction liquid at 917 cm⁻1 |
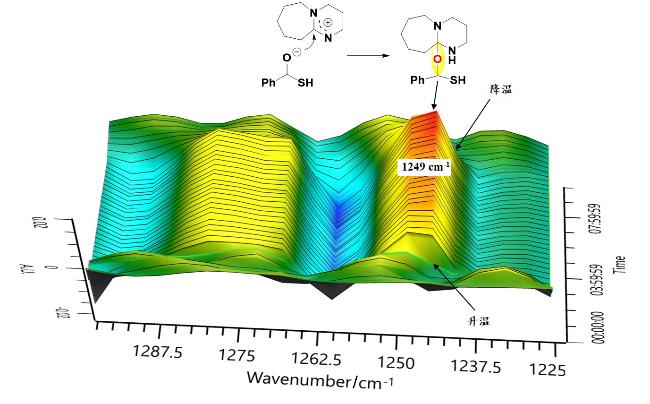
图3 反应液在1249 cm⁻1的原位红外光谱Figure 3 In situ FT-IR spectra of the reaction liquid at 1249 cm⁻1 |
2.5 反应机理推测
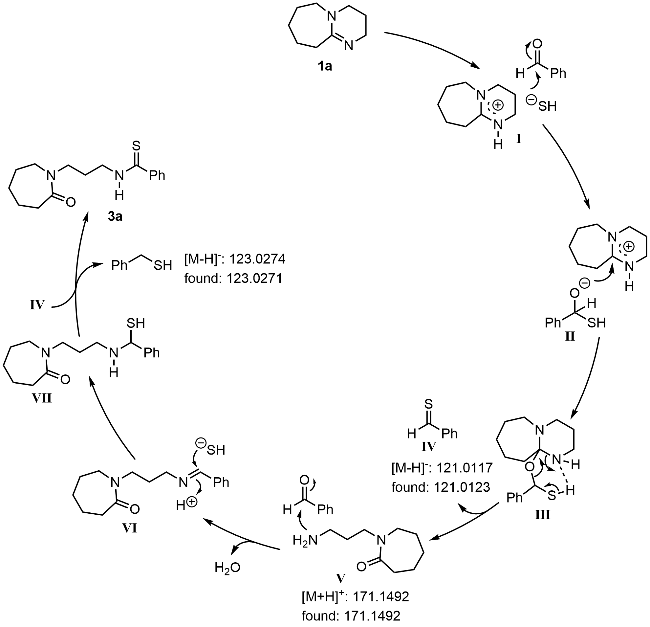
基于上述对照分析与原位红外结果, 对该反应机理进行合理推测(Scheme 6): 首先DBU (1a)与H2S形成中间体DBUH⁺HS⁻ (Ⅰ), 随后HS⁻亲核进攻苯甲醛, 形成含有O⁻离子的中间体(Ⅱ). 中间体(Ⅱ)继续亲核进攻DBUH⁺, 得到含有C—O—C键的中间体(Ⅲ), 该中间体通过原位红外光谱的特征信号峰得到了验证. 中间体(Ⅲ)通过分子内氢键作用发生[3,3]-σ迁移, 得到中间体(Ⅴ)和硫代苯甲醛(Ⅳ)(通过高分辨质谱(HRMS)检测得到), 中间体(Ⅴ)与苯甲醛继续反应, 形成亚胺中间体(Ⅵ). 随后HS⁻亲核进攻中间体(Ⅵ)形成中间体(Ⅶ). 最终, 中间体(Ⅶ)与中间体(Ⅳ)发生氧化还原反应形成目标产物3a和苄硫醇(通过HRMS检测得到).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
综上所述, 本研究基于环脒、醛类化合物与H2S的三组分协同反应体系, 成功开发了一种高效合成硫代酰胺衍生物的新方法. 该方法展现出优异的底物普适性, 特别是对含供电子基团的芳香醛展现出良好的反应活性, 成功拓展了硫代酰胺类化合物的结构多样性. 首次揭示了苯甲醛中氧原子作为亲核试剂触发CAH⁺开环并定向迁移至产物的新机制: 原位红外光谱动态捕捉到HS⁻亲核加成醛基形成的氧负离子进攻CAH⁺生成C—O—C关键中间体, 继而经分子内重排实现环脒开环, 最终成功合成了多种新型硫代酰胺类衍生物. 该策略实现了H2S资源化高效利用, 更为含硫功能分子的合成提供了新策略.
4 实验部分
将1 mmol DBU、2 mmol苯甲醛依次加入15 mL高压反应釜, 加入2 mL四氢呋喃, 放入磁子并把反应釜拧紧, 充入4 mmol H2S气体, 在130 ℃下搅拌反应24 h, 待反应结束后, 开釜用乙酸乙酯溶解反应液, 减压蒸馏除去溶剂, 得到粗产物. 再用200~300目硅胶进行柱层析分离(V(石油醚)/V(乙酸乙酯)=1∶1作为洗脱剂)得到纯度大于99%的目标产物.
(Cheng, B.)