

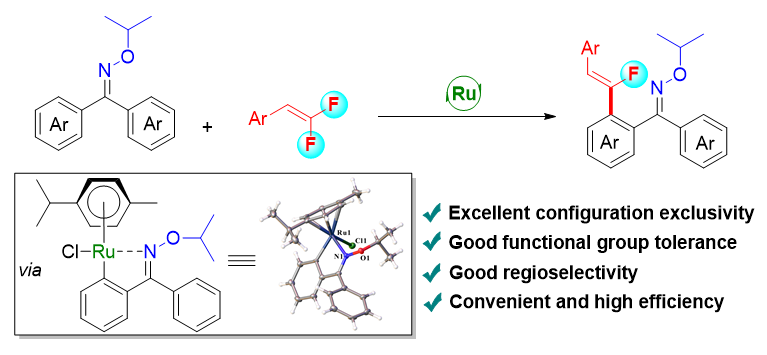
1 引言
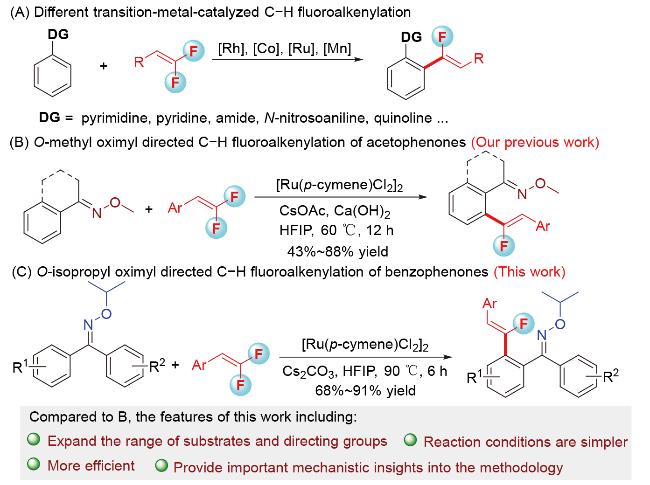
氟原子是目前应用最广泛的元素之一. 近年来, 合成化学家致力于将氟原子或含氟结构单元(如CF3、OCF3、SCF3、单氟烯烃等)引入母体化合物, 因为氟原子的引入能显著改变小分子的性质, 包括生物活性、代谢稳定性和亲脂性等[1]. 单氟烯烃在空间位阻和偶极特性方面与酰胺键相似, 常被用作药物开发中的生物电子等排体(图1)[2]. 目前, 过渡金属催化的C—H键活化已成为引入含氟烯烃的高效策略[3]. 2015年, Loh课题组[4]开创性地报道了Rh(III)催化嘧啶/吡啶导向的C—H键单氟烯化反应, 实现了芳(杂)环单氟烯烃的合成. 此后, Co(III)、Mn(I)和Ru(II)催化的单氟烯基化反应相继被报道(图2A)[5]. 同时, 多种含氮导向基团, 如酰胺[6]、N-亚硝基苯胺[7]和喹啉[8]被开发用于实现C—H单氟烯基化反应. 然而, 作为结构简单且易于安装的强效导向基团, 肟醚已在C—H活化/官能化领域(如酰胺化、芳基化、烯基化、不对称氢化)得到广泛应用[9], 但肟醚导向的C—H键单氟烯基化研究仍较为罕见. 2020年, 我们课题组[10]报道了一种甲氧基肟醚导向的苯乙酮C—H键单氟烯基化方法(图2B). 尽管该方法展现出良好的官能团耐受性和立体选择性, 但该反应的经济性、效率及机理明确性仍有待进一步提升.

2 结果与讨论
2.1 导向基筛选
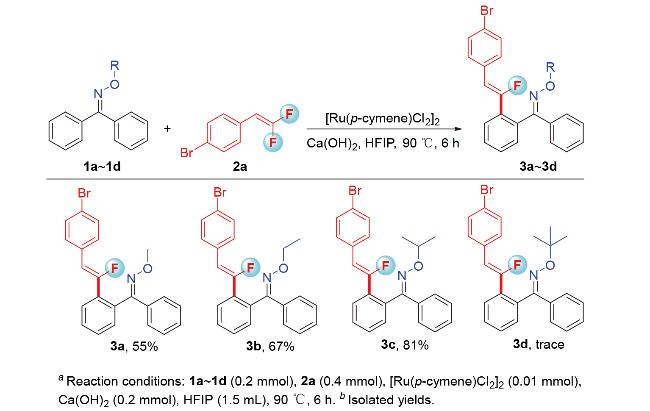
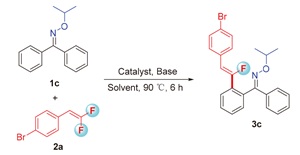
在90 ℃条件下, 将底物1a~1d (0.2 mmol)、2a (0.4 mmol)、[Ru(p-cymene)Cl2]2 (0.01 mmol)和Ca(OH)2 (0.2 mmol)置于1,1,1,3,3,3-六氟-2-丙醇(HFIP) (1.5 mL)中反应6 h. 结果表明, 当使用甲基肟醚、乙基肟醚和异丙基肟醚作为导向基时, 均能以单一Z构型获得目标产物, 其中异丙基肟醚表现出最高的反应效率(图3), 然而, 当采用叔丁基肟醚作为导向基时, 仅检测到痕量目标产物, 且薄层色谱(TLC)未观察到明显副产物, 其原因可能是叔丁基的存在不利于催化剂与肟醚氮原子配位. 因此, 最优导向基为异丙基肟醚.
2.2 反应条件优化
在确定最佳导向基团后, 我们对反应条件进行了系统评估(表1). 当使用[Cp*RhCl2]2、Pd(OAc)2和Cu(OAc)2作为催化剂或不添加催化剂时, 均未能获得目标产物(Entries 1~5). 令人欣喜的是, 在筛选多种常用碱后, 我们发现当使用Cs2CO3时, 3c的产率高达90% (Entries 6~10). 此外, 溶剂的选择对反应至关重要, 当改用2,2,2-三氟乙醇(TFE)作为溶剂时, 3c的收率骤降至34%; 而甲醇(MeOH)和四氢呋喃(THF)则完全无效(Entries 11~13). 与初始设定的反应温度相比, 将温度降至室温、60 ℃或升至100 ℃时均未能提高产率(Entries 14~16). 此外, 缩短反应时间至2 h会导致3c收率显著下降, 而延长至12 h则仅出现轻微降低(Entries 17, 18). 值得注意的是, 当碱用量减少至0.1 mmol时, 3c的收率急剧下降(Entry 19). 综合上述结果, 最终确定Entry 7所采用的条件为最佳反应条件.
表1 反应条件优化aTable 1 Optimization of the reaction conditionsa |
| Entry | Catalyst | Base | Solvent | Yieldb/% |
|---|---|---|---|---|
| 1 | [Ru(p-cymene)Cl2]2 | Ca(OH)2 | HFIP | 81 |
| 2 | [Cp*RhCl2]2 | Ca(OH)2 | HFIP | 0 |
| 3 | Pd(OAc)2 | Ca(OH)2 | HFIP | 0 |
| 4 | Cu(OAc)2 | Ca(OH)2 | HFIP | 0 |
| 5 | — | Ca(OH)2 | HFIP | 0 |
| 6 | [Ru(p-cymene)Cl2]2 | CsOAc | HFIP | 29 |
| 7 | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 90 |
| 8 | [Ru(p-cymene)Cl2]2 | DBU | HFIP | 0 |
| 9 | [Ru(p-cymene)Cl2]2 | pyridine | HFIP | 0 |
| 10 | [Ru(p-cymene)Cl2]2 | — | HFIP | 0 |
| 11 | [Ru(p-cymene)Cl2]2 | Cs2CO3 | TFE | 34 |
| 12 | [Ru(p-cymene)Cl2]2 | Cs2CO3 | MeOH | 0 |
| 13 | [Ru(p-cymene)Cl2]2 | Cs2CO3 | THF | 0 |
| 14c | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 16 |
| 15d | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 77 |
| 16e | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 84 |
| 17f | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 79 |
| 18g | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 89 |
| 19h | [Ru(p-cymene)Cl2]2 | Cs2CO3 | HFIP | 46 |
a Reaction conditions: 1c (0.2 mmol), 2a (0.4 mmol), catalyst (0.01 mmol), base (0.2 mmol), solvent (1.5 mL), 90 ℃, 6 h. b Isolated yields. c r.t. d 60 ℃. e 100 ℃. f 2 h. g 12 h. h Using 0.1 mmol Cs2CO3. DBU=1,8-diazabicyclo- [5.4.0]undec-7-ene. |
2.3 底物普适性考察
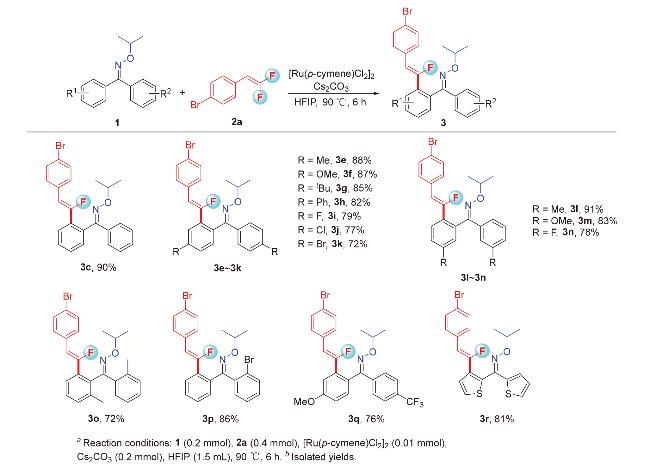
在确立最优反应条件后, 我们开始评估该催化体系下二苯甲酮肟醚的底物适用范围, 结果见图4. 总体而言, 该反应展现出优异的区域选择性, 能获得单一Z构型产物. 此外, 该方法对多种官能团表现出良好的耐受性, 无论是邻位、间位还是对位带有甲基(3e、3l、3o)、甲氧基(3f、3m)、叔丁基(3g)、苯基(3h)以及卤素基团(F、Cl、Br) (3i~3k、3n)的对称二苯甲酮肟醚, 均能以良好至优异的产率获得相应产物3e~3o. 值得注意的是, 该方法具有出色的位点选择性, 反应仅发生在空间位阻较小的位置(3l~3n). 此外, 含供电子基团的底物比吸电子基团底物能以更高收率获得目标产物, 且间位/对位取代的底物比邻位取代的底物表现出更强的反应活性. 另一方面, 该策略对非对称底物也展现出优异的区域选择性, 当使用电子偏性的底物1p和1q时, 该活化反应仅发生在电子云密度较高的芳香环上, 分别以86%和76%的收率得到产物3p和3q. 进一步地, 噻吩二酮这类杂环底物在该反应体系下也适用, 以81%的高收率生成目标产物3r.
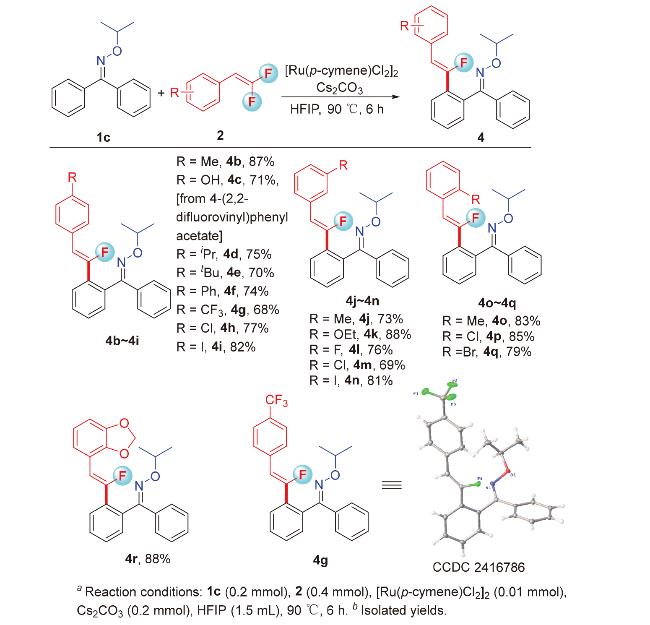
随后, 我们在标准条件下考察了偕二氟苯乙烯的底物适用范围(图5). 不出所料, 无论是邻位、间位还是对位带有供电子基团(如-Me, -OEt, -iPr, -tBu, -Ph)或吸电子基团(如-F, -Cl, -Br, -I, -CF3)的偕二氟苯乙烯, 都能适用该体系, 以68%~88%的收率得到相应的Z构型产物. 值得注意的是, 含有乙酸酯基的底物2c不仅能发生邻位C—H键单氟烯基化反应, 还能在Cs2CO3作用下脱去乙酸酯基, 以71%的收率生成脱酯产物4c. 为进一步确认产物构型, 我们通过单晶X射线衍射分析确定了产物4g的结构. 此外, 苯并[d][1,3]二氧杂环戊烯类偕二氟苯乙烯2r也能很好地兼容该反应体系, 以88%的收率得到产物4r.
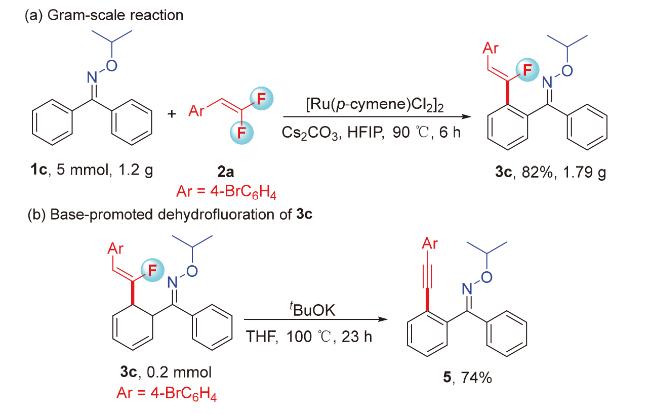
2.4 克级放大和应用研究
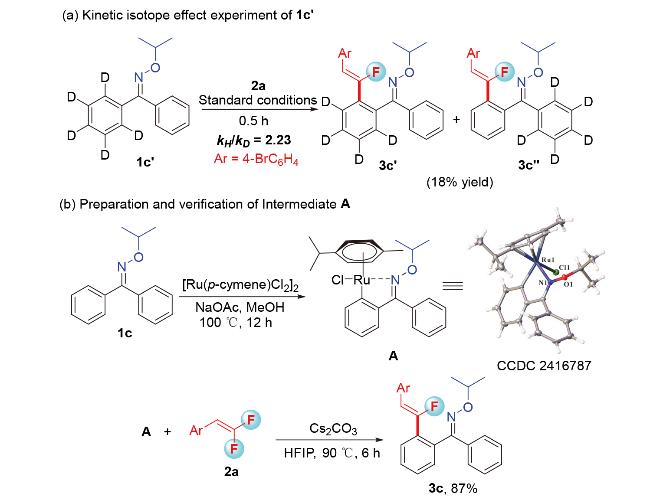
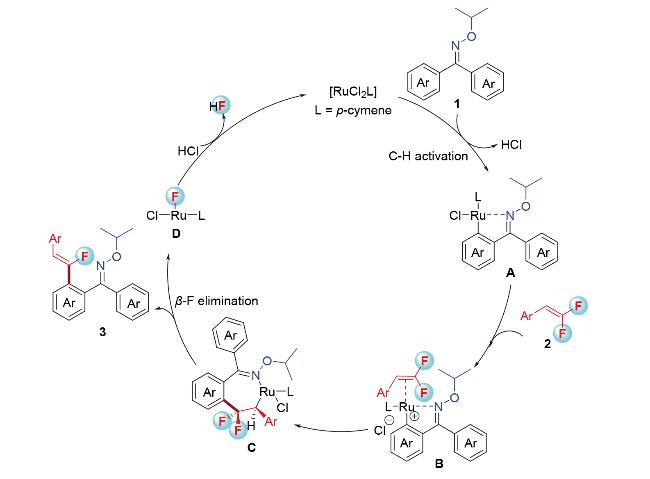
2.5 反应机理研究
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
总之, 我们开发了一种新型的Ru(II)催化的二苯甲酮肟醚类化合物Z-选择性C—H键单氟烯基化反应方法. 该策略具有以下显著特点: 优异的立体构型选择性、良好的区域选择性、广泛的底物适用范围、中等到优异的产率、可实现克级规模合成. 此外, 通过关键中间体A的成功确证, 进一步阐明了该反应的机理. 该方法有望为未来含氟药物的设计与发现提供一种高效工具.
4 实验部分
化合物3和4的合成: 将1 (0.2 mmol)、 加入2 (0.4 mmol)、[Ru(p-cymene)Cl2]2 (0.01 mmol)和Cs2CO3 (0.2 mmol)加入到10 mL玻璃密封管中, 向管中加入1.5 mL干燥HFIP, 拧紧封口塞后, 将封管置于90 ℃油浴中搅拌6 h. 反应结束后, 将反应混合物用30 mL二氯甲烷(DCM)稀释, 然后依次用水和盐水(各15 mL)洗涤, 收集有机相, 经无水硫酸钠干燥并过滤, 减压蒸除溶剂. 通过硅胶柱色谱纯化(洗脱剂: V(石油醚)/V(乙酸乙酯)=50∶1)得到产物3和4.
(Cheng, B.)