

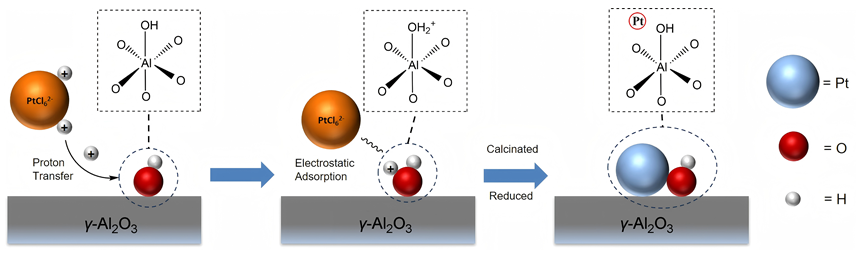
1 引言
2 结果与讨论
2.1 MA载体焙烧温度对MA负载Pt催化剂MCH脱氢催化活性的影响
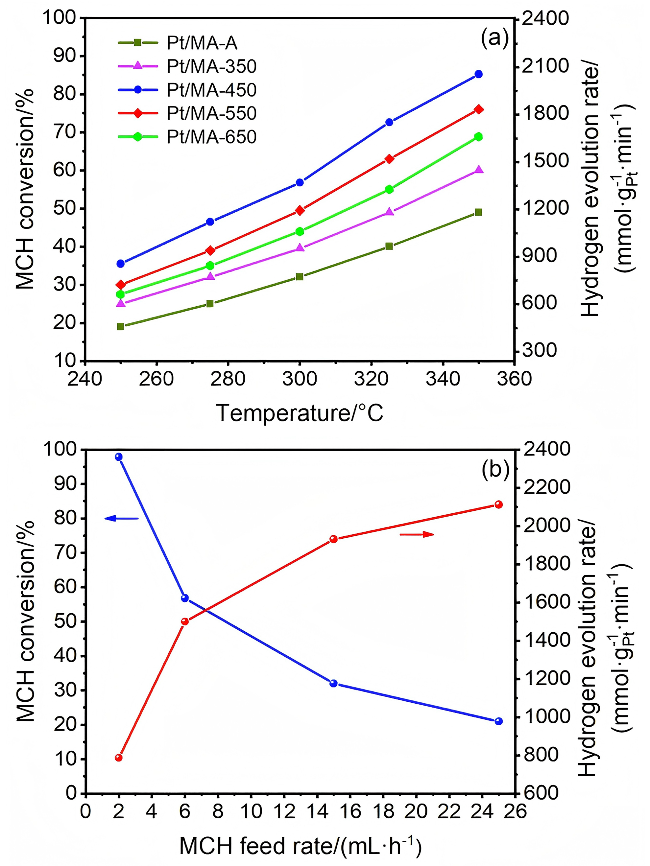
图1 (a)催化剂Pt/MA-T和Pt/MA-A在不同反应温度下的MCH转化率和相应的析氢速率及(b)催化剂Pt/MA-450在300 ℃不同MCH进料速率下的MCH转化率和析氢速率Figure 1 (a) MCH conversion and hydrogen evolution rate for catalysts Pt/MA-T and Pt/MA-A at different reaction temperatures and (b) MCH conversion and hydrogen evolution rate for catalyst Pt/MA-450 at 300 ℃ with different MCH feed rates |
表1 催化剂Pt/MA-450与文献报道的负载型Pt基催化剂MCH脱氢活性对比Table 1 Comparison of catalyst Pt/MA-450 with previous reported Pt-based catalysts for MCH dehydrogenation reaction |
| Catalyst | w(Pt)/% | Temperature/℃ | WHSV/h–1 | H2 evolution rate/(mmol•gPt–1•min–1) | Ref. |
|---|---|---|---|---|---|
| Pt/Mg-Al-O | 0.5 | 300/350 | 9.48 | 461.5/1892 | [18] |
| Pt/Al2O3 | 1.5 | 300 | 28.4 | 656 | [11] |
| B(Pt-n)/Al2O3 | 1 | 300 | 23.7 | 984 | [19] |
| 1%Pt/Clu-350 | 1 | 320 | 47.4 | 1118.8 | [20] |
| Pt1Sn6@S-1 | 0.4 | 350 | 75.84 | 1343 | [21] |
| Pt/SG-9 | 0.5 | 300 | 98.75 | 2081 | [15] |
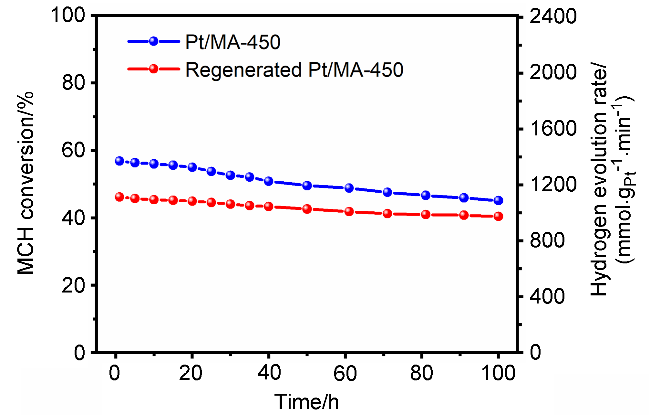
| Pt/MA-450 | 0.5 | 300 | 98.75 | 2112 | this work |
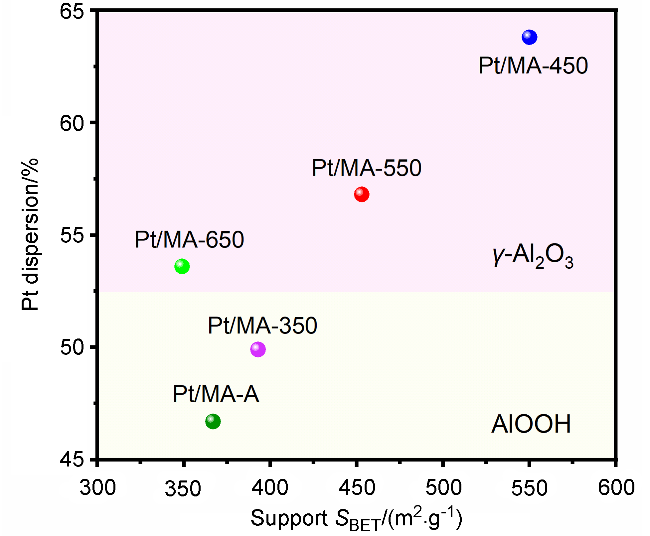
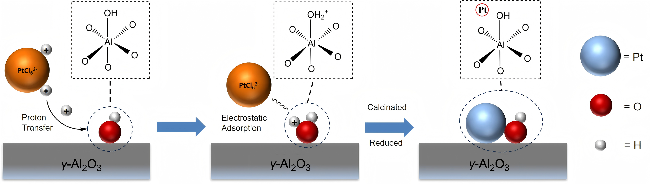
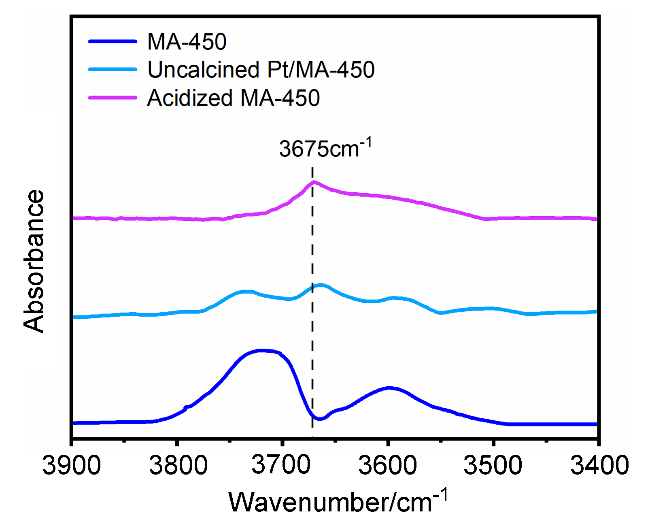
2.2 MA载体焙烧温度对Pt活性组分分散度的影响
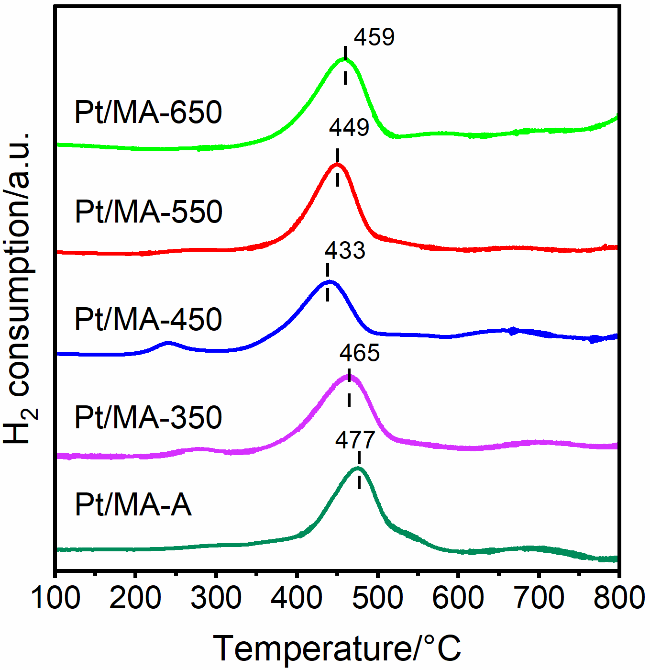
2.2.1 H2程序升温还原(H2-TPR)分析
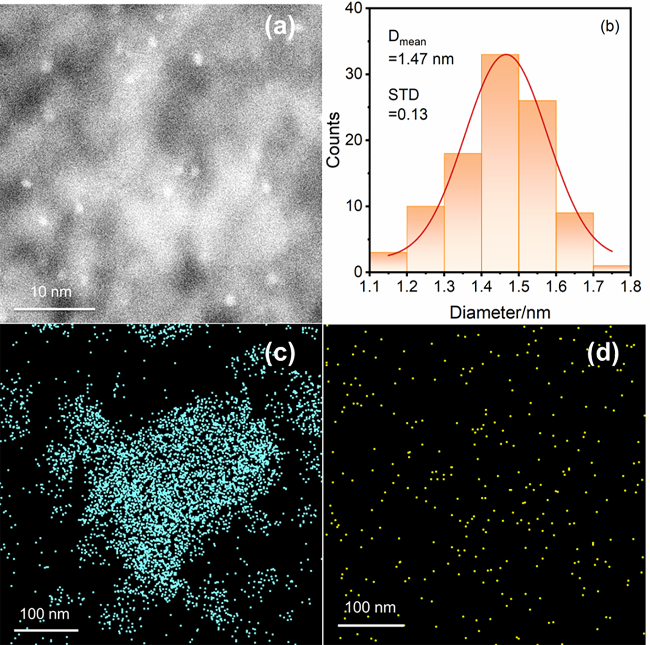
2.2.2 CO脉冲化学吸附分析和球差校正扫描透射电子显微镜(AC-STEM)表征
表2 催化剂Pt/MA-T和Pt/MA-A的CO脉冲化学吸附表征结果Table 2 CO pulse chemisorption characterization results for catalysts Pt/MA-T and Pt/MA-A |
| Catalyst | Dispersion/% | Particle size/nm | Metal surface area/ (m2•g–1) |
|---|---|---|---|
| Pt/MA-A | 46.7 | 2.05 | 116.8 |
| Pt/MA-350 | 49.9 | 1.89 | 123.1 |
| Pt/MA-450 | 63.6 | 1.49 | 157.1 |
| Pt/MA-550 | 56.8 | 1.66 | 140.1 |
| Pt/MA-650 | 53.6 | 1.76 | 132.3 |
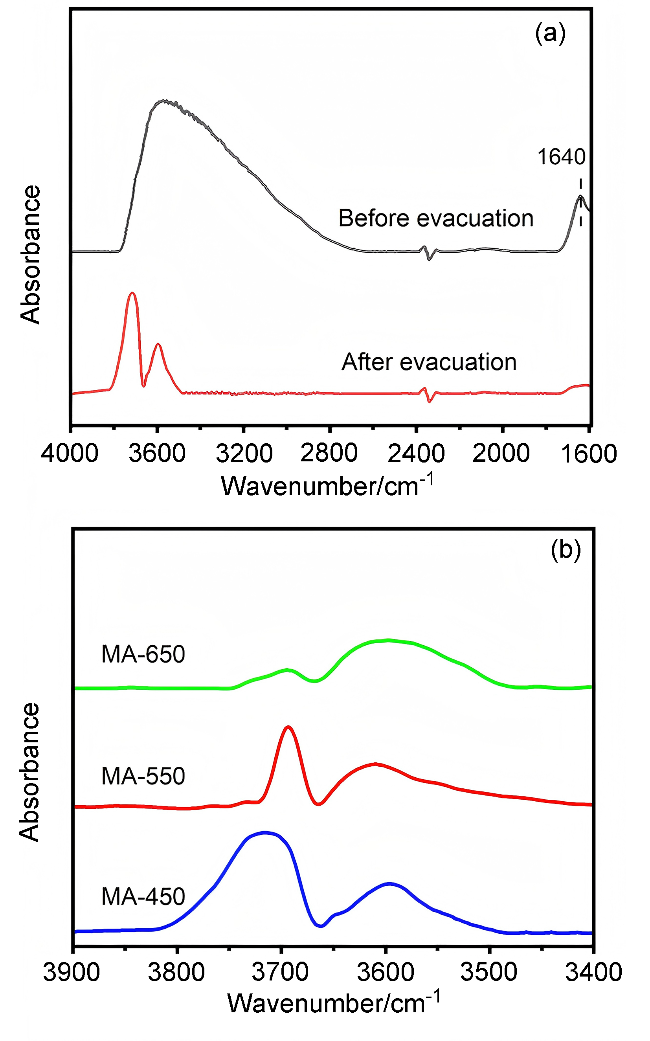
2.3 焙烧温度对MA载体性质的影响
2.3.1 N2物理吸附-脱附(BET)分析
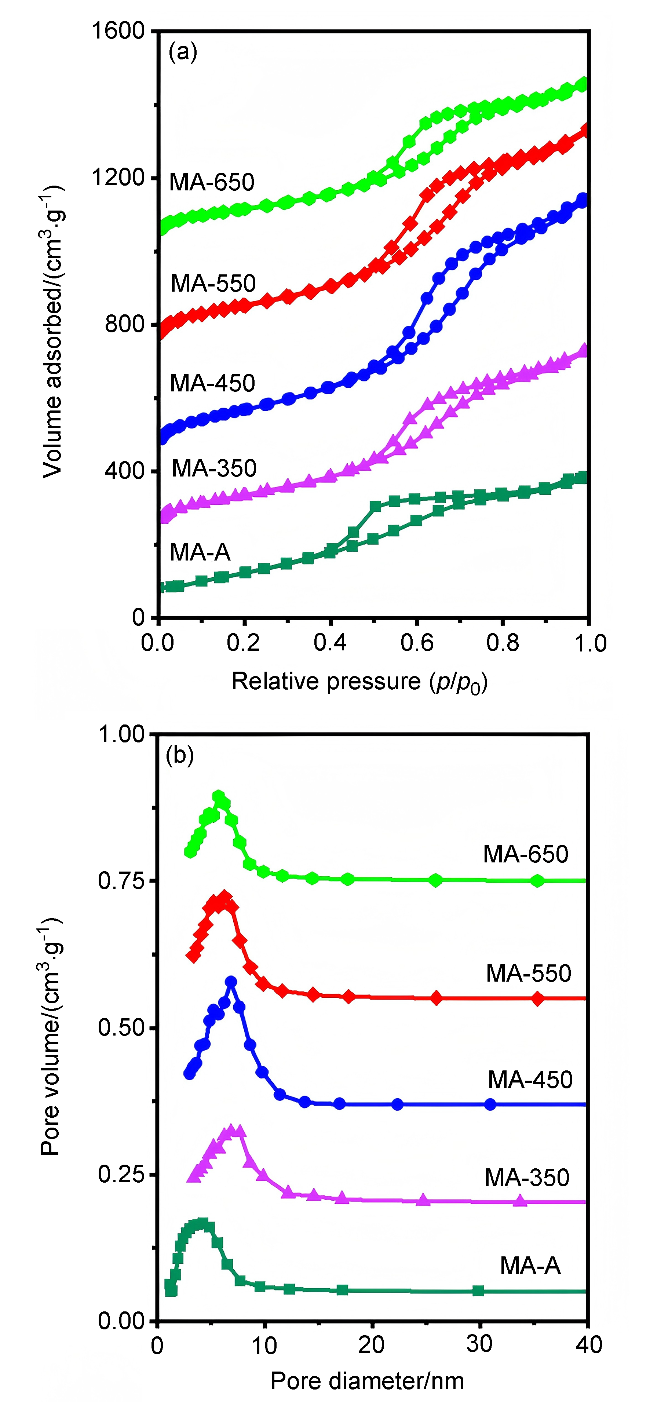
图4 载体MA-T和MA-A载体MA-T和MA-A的(a) N2吸附-脱附等温线及(b)相应的孔径分布曲线 Figure 4 (a) N2 adsorption-desorption isotherms and (b) the corresponding pore size distribution curves of supports MA-T and MA-A |
表3 载体MA-T和MA-A的比表面积、孔体积和孔径Table 3 Specific surface areas, pore volumes, and pore sizes for supports MA-T and Pt/MA-A |
| Support | SBET/(m2•g–1) | Vp/(cm3•g–1) | Dp/nm |
|---|---|---|---|
| MA-A | 367 | 0.55 | 4.34 |
| MA-350 | 401 | 0.75 | 6.93 |
| MA-450 | 550 | 1.21 | 7.02 |
| MA-550 | 453 | 0.95 | 6.78 |
| MA-650 | 349 | 0.61 | 5.81 |
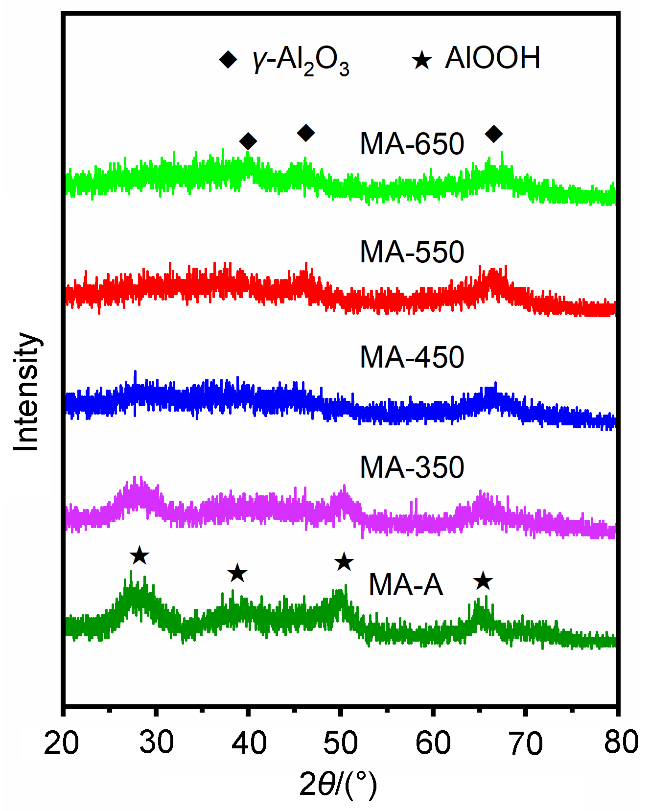
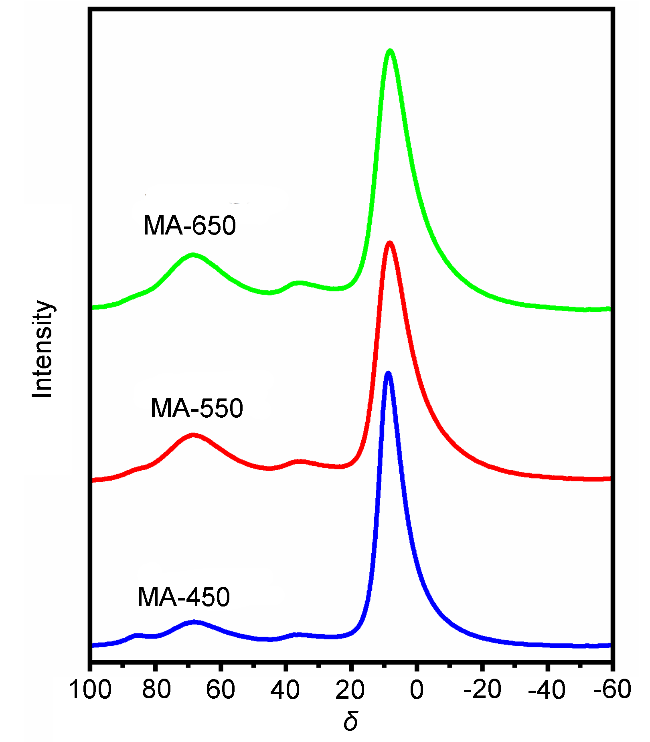
2.3.2 X射线衍射(XRD)分析
2.3.3 X射线光电子能谱(XPS)分析
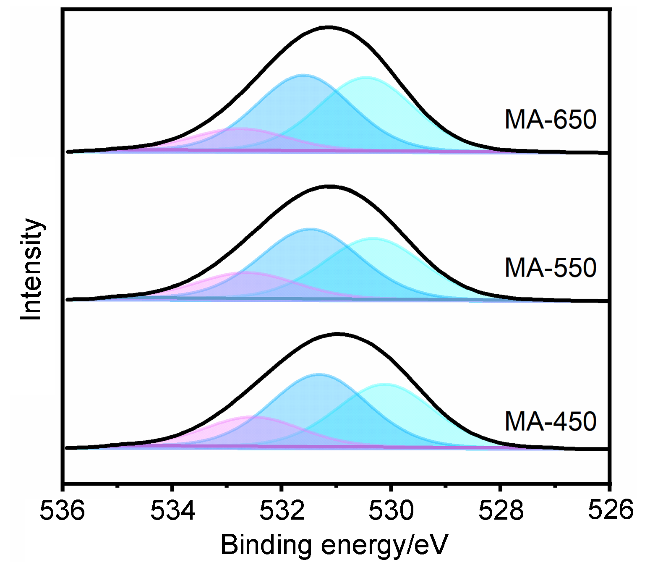
图7 载体MA-450、MA-550和MA-650的O 1s XPS图谱Figure 7 O 1s XPS spectra of supports MA-450, MA-550, and MA-650 |
表4 载体MA-450、MA-550和MA-650表面元素组成及各种表面氧物种的相对含量Table 4 Composition of surface element measured by XPS analysis and proportions of different surface oxygen species for supports MA-450, MA-550, and MA-650 |
| Support | x(O)/% | x(Al)/% | x(OL)/% | x(OV)/% | x(OH)/% |
|---|---|---|---|---|---|
| MA-450 | 55.39 | 29.81 | 21.16 | 24.32 | 9.91 |
| MA-550 | 54.76 | 30.37 | 21.29 | 24.43 | 9.04 |
| MA-650 | 54.93 | 30.83 | 22.41 | 24.88 | 7.64 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}