


1 引言
随着全球能源需求的持续增长和环境问题的日益严峻, 建设以新能源为主体的新型电化学储能系统逐渐成为社会的焦点[1]. 锂离子电池(Lithium-ion batteries, LIBs)作为技术成熟的新型电化学系统之一, 在便携式电子设备和电动汽车市场取得了巨大的成功[2-4]. 但由于锂资源的匮乏以及分布不均等问题, 使得锂离子电池的使用成本提高, 且容易出现我国锂资源被卡脖子的问题, 因此开发低成本、高性能的储能系统显得十分重要. 钠离子电池(Sodium ion batteries, SIBs)由于钠资源丰富、成本低廉、安全性好, 且具有与锂离子电池相似的电化学储能机制, 其在大规模储能应用中具有极大的潜力[5-7].
正极材料作为SIBs的重要组成部分之一, 不仅直接影响电池关键性能指标, 也是决定SIBs成本的重要因素[8-9]. 在过去的几十年里, SIBs正极材料已经相继取得一些重要进展[10]. 目前钠离子电池的正极材料主要可以分为层状过渡金属氧化物、普鲁士蓝类似物以及聚阴离子型化合物三大类. 层状过渡金属氧化物因其高比容量、高电压平台而被认为是极具潜力的正极材料[11]. 然而该类材料在充放电过程中会经历不可逆的相变, 易导致结构破坏和电化学性能下降, 并且其空气稳定性较差, 增加了储存成本[12]. 对于具有三维开放骨架结构的普鲁士蓝类似物来说, 其宽敞的离子通道有利于钠离子可逆的嵌入脱出, 通常具有较高的理论比容量[13]. 但其合成过程中容易产生结晶水和晶格缺陷, 限制了在钠离子电池中的大规模应用. 与前两种正极材料相比, 聚阴离子型化合物不仅具有稳定的三维框架结构, 有利于钠离子快速嵌入脱出, 并且在钠离子脱嵌过程中体积变化小, 提供了优异的稳定性和安全性[14]. 同时聚阴离子强烈的诱导效应可以调节过渡金属氧化还原电对的能 量[15], 提高其工作电压, 进而保证较高的能量密度.
近年来研究者们聚焦于Na3V2(PO4)3[16]、Na4VMn(PO4)3[17]等钒基和锰基聚阴离子型正极材料, 虽然在电化学性能方面取得了重要进展, 但有毒元素钒的存在以及锰的Jahn-Teller效应限制了它们的规模应 用[18]. 铁基聚阴离子型正极材料同时具有成本低廉、环境友好和结构稳定等优点, 被认为是钠离子电池中极具潜力的正极[19-20]. 铁基硫酸盐正极材料凭借相对较低的原材料成本和较高的工作电压脱颖而出. Na2Fe(SO4)2[21]、Na2Fe2(SO4)3[22] (NFS)和Na2Fe(SO4)2• nH2O[23]等铁基硫酸盐材料自2014年被相继提出作为钠离子电池正极材料后得到了广泛的研究. 得益于SO2- 4强烈的诱导效应, 这些铁基硫酸盐均表现出较高的工作电压, 其中NFS材料的工作电压更是高达3.8 V (vs. Na+/Na). Na2Fe(SO4)2•nH2O采取的是伪层状单斜排列结构, 这种结构不利于长期循环的稳定性, 而Na2Fe(SO4)2和NFS具有聚阴离子型材料典型的稳定3D框架, 有利于Na+的快速脱嵌. 不仅如此, Na2Fe(SO4)2和Na2Fe(SO4)2•nH2O较低的理论比容量导致它们在实际应用中竞争力受限. 因此综合来看, 具有最高的理论比容量(120 mAh/g), 理论能量密度高达456 Wh/kg, 并且结构稳定、成本低且环境友好的NFS材料是一类具有很好应用前景的钠离子电池正极材料. 但值得注意的是, 该材料本征电导率低、界面稳定性差, 易吸水, 并且合成过程容易产生杂质, 难以获得纯相材料, 距离规模商业应用仍面临巨大挑战. 目前已有大量的研究工作致力于解决NFS材料的上述关键问题, 助力推进其产业化进程, 但学术界仍缺乏对该材料产生这些问题的本质原因以及解决措施的系统性总结. 本文将从NFS的晶体结构和钠离子脱嵌机制出发, 系统性概述其面临的关键问题以及改进策略, 并总结其产业化进程, 旨在为广大科研工作者们提供设计策略, 推动NFS材料的研究和应用进程.
2 硫酸铁钠的性质
2.1 晶体结构
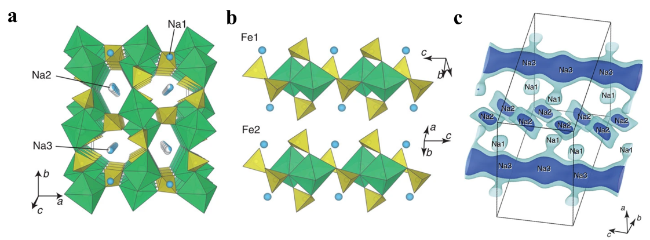
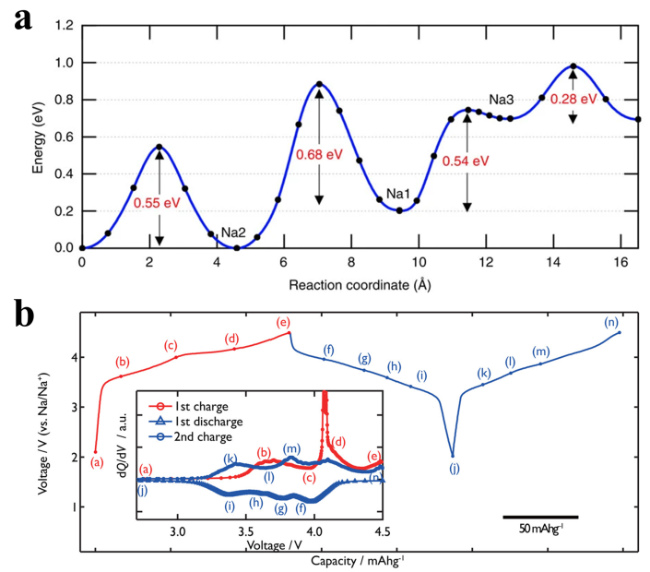
NFS晶体结构如图1a所示, 与大部分采用NASICON相关结构的AxM2(XO4)3型化合物不同, NFS不含灯笼单元[M2(XO4)3], 形成了具有Alluaudite型框架的独特结构, 为C2/c对称的单斜晶格, 通式可以表示为AA′BM2(XO4)3, 其中A=Na(2), A′=Na(3), B=Na(1), M=Fe2+, X=S. 在这种结构中, 一对等效的FeO6八面体形成Fe2O10二聚体, 并与SO2- 4四面体通过共角连接, 沿c轴形成稳定的3D结构. 如图1b所示, 在Fe2O10二聚体单元中, Fe离子占据了两个不同的晶体点位, 即Fe(1)和Fe(2). 尽管Fe(1)和Fe(2)的局部结构相似, 但Mõssbauer光谱揭示了它们在晶体学上是不同的, 并且这种独特的局部Fe配位结构使得Fe-Fe原子之间的距离仅为0.32 nm, 这为实现高氧化还原电位提供了帮助. 而对于NFS结构中的三个Na位点, 只有Na(1)位点是被完全占据的, Na(2)和Na(3)位点均是被部分占据, 并且键价(Bond valence, BV)方法分析显示对于NFS材料仅存在沿c轴的一维扩散通道(图1c). 密度泛函理论(Density functional theory, DFT)计算了Na+在三个不同位点结合能和迁移活化能[22]. 图2a中显示Na与Na(2)位点的结合能最低, 而与Na(3)位点的结合能最高. 并且可以发现, Na+在Na(3)通道内的迁移能是最低的, 仅为0.28 eV, 这说明Na+迁移优先发生在该结构中的Na(3)位点, 展现出优异的离子迁移动力学. 对于通道间的迁移, Na+在Na(1)和Na(2)位点之间的迁移活化能为0.88和0.58 eV, 而在Na(1)和Na(3)之间的迁移活化能仅为0.54和0.05 eV, 这使得Na+在Na(1)和Na(3)位点之间能够轻易地进行迁移. 因此可以认为这种材料在Na(2)和Na(3)位点沿c轴具有一维Na+传输通道, 而Na(1)位点的Na+可以通过Na(3)位点提取, 这使得所有Na+都可以用于脱嵌反应, 对理论容量没有限制.
2.2 钠离子脱嵌机制
NFS材料的充放电曲线和相应的差分恒流曲线(dQ/dV)如图2b所示, 可以看见dQ/dV曲线在第一次充电过程显示出了3.65 V和4.06 V两个氧化峰, 而在第一次放电过程于3.42、3.80、4.04 V处显示出了三个还原宽峰. 第一次充电过程中3.65 V的氧化峰对应着Na从Na(3)位点的脱出, 这与Na(3)位点最高Na+结合能的计算结果保持一致. 紧接着Na从Na(1)位点和Na(2)脱出, 并诱导Fe3+从Fe(1)位点迁移到空的Na(1)位点, 从而表现出4.06 V处的氧化峰. 而第一次放电过程中, 初始放电的4.04 V的还原峰归因于Na在Na(1)和Na(2)位点的嵌入. 在较深放电(3.80 V)中Na开始嵌入Na(3)位点, 放电最后一步(3.42 V)则对应着Na嵌入空的Fe(1)轨道[27]. 随后的第二次充电过程, dQ/dV曲线出现了三个氧化峰, 且位置明显左移, 同时初始充电的电压曲线也与后续循环的电压曲线略有不同, 这说明在第一次脱钠过程中, NFS发生了一些不可逆的结构转变, 类似于Li2FeSiO4[28-29]和Li2FeP2O7[30-31]的情况. Oyama等[26]利用X射线晶体衍射(X-Ray Diffraction, XRD)和价键总和计算证实了第一次脱钠过程中的不可逆结构转变是由于dQ/dV曲线在4.06 V处时, Na从Na(1)位点脱出, Fe2O10二聚体中强大的库伦斥力诱导Fe3+从Fe(1)位点迁移到空的Na(1)位点导致的, 但这种Fe迁移只发生在第一个充电过程中, 随后循环表现出高度的Fe3+/Fe2+氧化还原可逆性(体积变化仅为3.5%), 这也保证了NFS材料的稳定性.
3 硫酸铁钠正极材料面临的问题
3.1 纯相结构难以获得
获取纯相NFS材料对于充分发挥其电化学性能具有十分重要的意义, 然而在NFS材料的合成过程中, 人们发现由于Fe2O10二聚体中Fe和Fe之间较大的库仑斥力造成Fe2+逃逸, 产物中总是会存在非化学活性的α/β-FeSO4、Fe3O4和Na6Fe(SO4)4等杂质, 使实际的比容量明显低于理论比容量. 目前大量研究表明, Na占据Fe位可以缓解Fe2O10二聚体中Fe之间的库仑斥力, 降低杂质含量, 并最终以Na2+2xFe2-x(SO4)3 (NFSx)这种非化学计量比的形式存在, 因此合理设计结构中的Na/Fe比是获得纯相NFS材料的关键.
3.2 本征电导率低
与大部分聚阴离子型材料相同, NFSx晶胞中过渡金属离子被不传导电子的聚阴离子SO2- 4基团分隔, 其价电子的电子云被孤立阻碍了电子交换, 材料的本征电子电导率低. Lu等[32]利用恒电位极化和电化学阻抗谱测试了NFSx的离子和电子传输速率. 虽然NFSx表现出较高的离子电导率(1×10−7 S/cm), 但其电子电导率仅为2×10−9 S/cm, 这使得NFSx在实际应用时倍率性能较差, 电化学性能难以发挥. 为了促进NFSx材料的应用, 需要通过元素掺杂、碳层包覆等改性手段来提高其电子电导率.
3.3 界面稳定性差
尽管SO2- 4基团强烈的诱导效应为NFSx材料提供了高电压平台, 但同时强极性的SO2- 4基团使得其表面具有较高的电子密度, 对极性小分子(H2O、电解液中的酯类)具有高吸引力, 从而造成NFSx材料暴露在空气中不仅容易与环境中的水反应变成水合相Na2Fe(SO4)2•4H2O而损害材料的结构, 也可能在电极/电解质界面处引发严重的副反应[33].
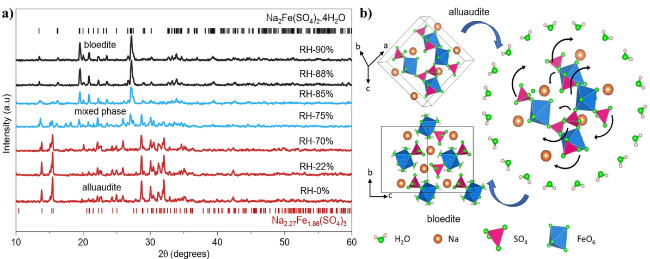
虽然水合相Na2Fe(SO4)2•4H2O具有一定的化学活性, 但其较低的理论比容量和较大的内阻会显著降低NFSx材料的电化学性能. Barman等[34]计算了Na2SO4- FeSO4二元组成的生成焓, 发现水合相Na2Fe(SO4)2• 4H2O的生成焓(-117.16 kJ/mol)远低于NFSx (-11.76 kJ/mol), 这说明水合相Na2Fe(SO4)2•4H2O比NFSx在热力学上更稳定. 为了进一步探究NFSx材料暴露在空气中的结构演变, 他们将制备的Na2.27Fe1.86(SO4)3置于不同相对湿度(RH)的环境下暴露1 h, 并进行了XRD测试(图3a). 当相对湿度高于70%时, NFSx的衍射峰逐渐消失, 并出现Na2Fe(SO4)2•4H2O的衍射峰, 直到相对湿度高于85%时, NFSx完全转化成Na2Fe(SO4)2•4H2O. 图3b展示了这种水分侵蚀的过程. 总而言之, NFSx材料在合成后需要小心存放在惰性气氛中, 以保证其电化学活性.
降低NFSx材料表面电子密度是提高其界面稳定性的关键, 较高的表面电子密度使得NFSx材料组装成电池运行时在高电压下会发生溶剂的分解, 导致形成不稳定和不均匀的CEI膜, 从而阻碍Na+在电极界面的迁移, 并导致电极界面发生连续的副反应, 最终导致电化学性能的快速衰退[35].
4 硫酸铁钠材料的合成方法
NFS材料的合成面临着两个挑战: (1)在水性介质中溶解; (2)在450 ℃以上分解. 因此它对合成条件较为苛刻. 目前主要可以分为固相法(球磨法)和液相法两大类, 其中液相法具体包括喷雾干燥、冷冻干燥法和离子热法等.
4.1 固相法
固相法是目前制备硫酸铁钠的主流方法, 该方法合成步骤简单, 通常将硫酸钠、硫酸亚铁等原料进行球磨充分混合后高温煅烧即可得到NFS材料. 固相法在球磨前通常需要对FeSO4•7H2O原料进行热处理脱水, 避免原料中的结晶水影响球磨过程以及侵蚀产物. 并且由于硫酸铁钠材料较低的本征电导率, 在球磨时通常会加入无机碳源, 因而固相法得到的材料通常容量较高. Zhu等[36]通过固相法与科琴黑简单混合后获得的NFS材料初始可逆容量可达85.3 mAh/g. 但固相法得到的NFS材料通常粒径较大、粒度分布不均, 且无法得到特定形貌的材料.
4.2 液相法
相较于固相法, 液相法通常能实现原子级别的混合, 并且容易制备出微米或纳米级产物. 目前NFS材料使用较为广泛的液相法主要包括喷雾干燥法、冷冻干燥和离子热法.
喷雾干燥法由于其高效、低能耗的优点而广泛应用于材料的规模制备. 过去研究者们普遍认为由于NFS材料吸水变质的特性, 喷雾干燥法并不适用于NFS材料的合成. 直到近几年, Barman等[34]提出喷雾干燥法可以通过中间相Na2Fe(SO4)2•4H2O的形成获得NFS产物. 他们首先将原料溶于水后, 通过第一步喷雾干燥形成中间相Na2Fe(SO4)2•4H2O(式(1)), 随后通过第二步煅烧过程转化为所需的NFS材料(式(2)), 其初始容量为77 mAh/g. 与固相法相比, 喷雾干燥不需要长时间的球磨和去除原料结晶水的过程, 可以明显提高生产效率. 其制备的NFS材料不仅粒径小、粒度相对均一, 并且可以制备多孔/中空微球.
Na2SO4+2FeSO4•7H2O→Na2Fe(SO4)2•4H2O+FeSO4+10H2O
Na2Fe(SO4)2•4H2O+FeSO4→Na2Fe2(SO4)3+4H2O
冷冻干燥法是一种通过先冷冻后升华来脱除物料中水分的工艺, 冰晶升华后留下丰富的孔隙, 可以形成高比表面积的多孔网络结构. 其合成过程中的低温和真空特性可以有效防止NFS材料分解或氧化, 从而得到纯度较高的产物. Chen等[37]通过冷冻干燥法合成的NFS微/纳米颗粒表现出优异的电化学性能, 在0.1 C下放电比容量高达107.9 mAh/g. 但冷冻干燥法所需时间长, 设备投入较高, 且目前工业级连续冻干技术尚不成熟, 难以实现连续化生产.
离子热法可以合成特定形貌和尺寸的材料, 对于NFS这种仅沿c轴具有一维Na+传输通道的材料来说, 通过特定形貌设计减小沿c轴粒径对于提高其电化学性能具有一定效果. 离子热法通常是将原料分散在装有离子液体(例如1-乙基-3-甲基咪唑鎓双(三氟甲磺酰基)酰亚胺[38])的高压反应釜中, 在高温高压下保持若干小时后进行离心、干燥和煅烧得到所需的NFS材料. 离子热法得到的NFS材料通常具有纳米粒径, 并且纯度高, 但其需要使用价格昂贵的离子液体且对设备要求较高, 不利于规模化生产.
4.3 其他合成方法
NFS材料在水性介质中溶解和高温分解的特性使得其对合成方法和条件要求非常高. 针对性开发新的合成方法对于得到纯度高, 且形貌和粒径可控的高性能NFS材料显得十分重要.
Li等[39]借鉴了纳米材料在油酸(OA)、油胺(OAm)等高沸点有机溶剂中的合成, 开发了一种高沸点有机溶剂辅助胶体合成(HOS-CS)和烧结工艺结合的方法来获得纳米级NFS材料. 他们使用OA和OAm混合物作为溶剂和封端剂, 使HOS-CS过程仅需30 min便完成了反应物向尺寸和元素分布均匀的前驱体转化. 并且OA和OAm在随后的烧结工艺中可以原位转化为碳源. 最终获得的纳米级NFS材料具有优异的倍率性能, 即使在30 C倍率下仍能提供46.9 mAh/g的放电比容量.
Yang等[40]注意到喷雾干燥法的产物需要高温煅烧水合相Na2Fe(SO4)2•4H2O转化为NFS材料, 而从加热的结晶水中释放的蒸汽可能会破坏NFS晶体的完整性, 并可能通过水合反应将其还原为Na2Fe(SO4)2•4H2O材料, 因此他们提出一种通过无水Na2Fe(SO4)2前驱体高温煅烧转化为NFS材料的合成路线: 通过溶胶-凝胶法得到水合相Na2Fe(SO4)2•4H2O, 并在200 ℃下真空干燥去除结晶水形成无水Na2Fe(SO4)2前驱体, 随后煅烧使其转化为NFS材料. 相较于水合相Na2Fe(SO4)2•4H2O直接煅烧得到的NFS材料, 通过无水Na2Fe(SO4)2煅烧得到的NFS材料表现出更高的相纯度和结构稳定性(2 C下循环200次容量保持率超过95%).
5 硫酸铁钠正极材料的改性手段
目前对NFS材料改性手段的主要目的是优化电子/离子传输、降低表面电荷密度以及缓解晶体结构内部斥力, 旨在提高其相纯度、本征电导率以及界面稳定性, 从而促进其电化学性能的发挥. 具体改性手段如下.
5.1 非化学计量比设计
Oyama等[41]研究发现在前驱体混合物中添加适当过量的硫酸钠使得部分Na取代晶体结构中的Fe位可减少杂质含量, 这表明非化学计量比的富钠相NFSx的设计可以提高材料的纯度. 他们系统研究了Na2SO4-FeSO4体系的相平衡, 采用球磨固相法合成了一系列NFSx (0≤x≤0.4)材料. XRD测试表明非化学计量比的NFSx与NFS同构型, 均属于Alluaudite结构. 当x=0时, 产物中α/β-FeSO4和Fe3O4杂质含量超过了15% (w); 而当x=0.25时, 产物中α/β-FeSO4和Fe3O4杂质含量下降到了1.2% (w); 进一步增加到0.3以上时, 晶格参数变化很小, 并且出现了新的富钠相杂质Na6Fe(SO4)4. 这些结果表明合成的NFS在此条件下并不稳定[21], 而是趋向于形成NFSx, 其中x约为0.25~0.3, 进一步通过Fe的Mõssbauer光谱确定了准确组成为Na2.56Fe1.72(SO4)3.
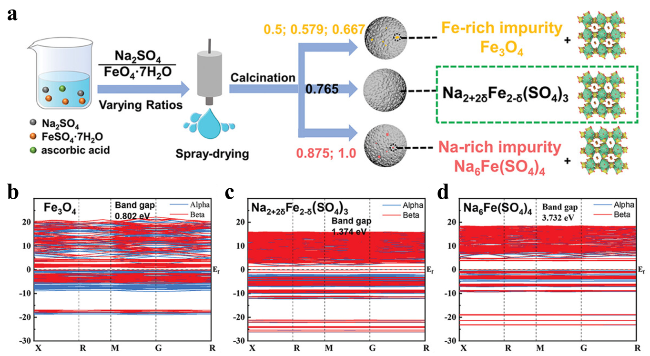
Yang等[42]也通过调节Na2SO4-FeSO4•7H2O体系的比例, 系统地研究了NFSx和杂质之间的非平衡演化机制. 图4a展示了他们的合成路线, 通过设计原料中n(Na2SO4)/n(FeSO4•7H2O)=0.5、0.579、0.667、0.765、0.875、1.0, 采用喷雾干燥法合成了一系列NFSx材料. XRD的Rietveld精修发现仅当Na2SO4/FeSO4•7H2O=0.765时获得的产物不存在杂质, 当比例减小时产物中出现富铁杂质Fe3O4, 而当比例增加时则出现富钠杂质Na6Fe(SO4)4. 为了评估杂质对NFSx材料电化学性能的影响, 作者采用DFT计算了富铁杂质Fe3O4、NFSx材料和富钠杂质Na6Fe(SO4)4的能带结构和带隙(图4b~4d). 与NFSx的带隙1.374 eV相比, 富钠杂质Na6Fe(SO4)4的带隙较大, 为3.732 eV. 带隙的增加表明电子跃迁到导带需要更高的能量, 意味着较差的电子电导率. 富铁杂质Fe3O4的带隙虽然仅为0.802 eV, 然而其在充放电过程中不能储存Na+, 意味着较差的离子电导率[43]. 因此, 这些杂质的存在会降低材料电化学性能. 电化学测试显示当比例为0.765时, 目标产物表现出最高的比容量(93.8 mAh/g)和最低的电荷转移电阻(239.1 Ω).
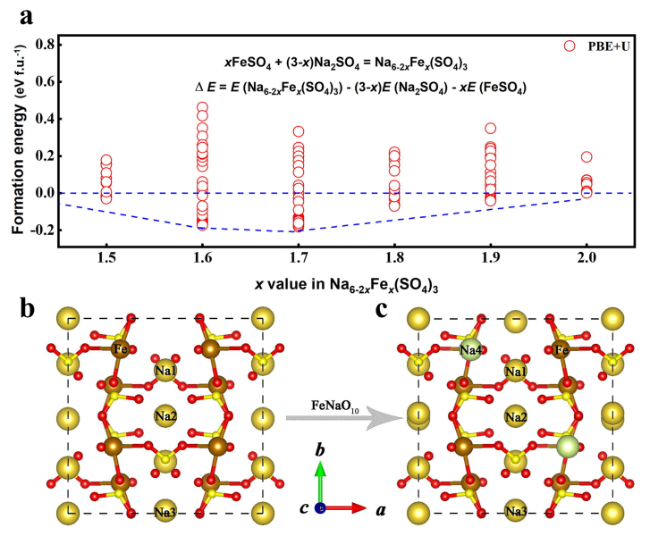
以上结果显示, 通过设计非化学计量比NFSx材料, 能够有效降低所得产物中的杂质含量, 从而优化材料的电化学性能. 因此, 明晰非化学计量比NFSx的内在形成原因对于设计高性能钠离子电池正极材料具有十分重要的意义. Zhao等[44]采用球磨法合成了Na6-2xFex(SO4)3 (1.5≤x≤2.0) (NFSO-x), 并通过DFT计算了一系列NFSO-x结构的形成能(图5a). 所有非化学计量比的富钠材料结构均比初始NFSO-2.0结构具有更低的形成能, 表明Na在结构中取代Fe位有利于提高结构稳定性. 他们提出造成这种现象的原因是Fe2O10二聚体中Fe-Fe原子距离较短, 较大的库仑排斥力使得结构极不稳定, 而FeNaO10二聚体的形成可以缓解结构中的库仑斥力, 提高结构稳定性(图5b、5c). 同时他们还发现在首次充电过程中Fe迁移到Na(1)位引起的不可逆结构转变也是由Fe2O10二聚体中的强库仑斥力引起的, 但这种不可逆结构转变有助于结构材料的稳定, 且不会影响材料的电子电导率. 最终, 具有最低形成能的NFSO-1.7材料表现出最佳的电化学性能, 在0.1 C下放电比容量为104.5 mAh/g, 而NFS(NFSO-2.0)放电比容量仅为59.9 mAh/g.
5.2 元素掺杂
5.2.1 阳离子掺杂
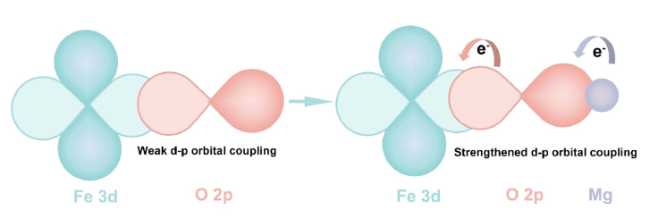
在过渡金属Fe位点引入具有不同电子态和半径的阳离子可以有效改变材料的电子结构和晶胞大小, 实现电导率和Na+迁移速率的提高. Zhou等[52]将镁离子掺入到FeO6结构中来调节NFSx的电子状态. Mg的引入使得Fe的3d轨道上的电子态发生了重新排布, 在费米能级附近出现了新的电子态, 从而使得掺杂后材料(Mg-NFS)的带隙从2.32 eV减小到2.16 eV. 低电负性的Mg掺入到FeO6结构中会促进更多的电子向桥连O原子(Fe-O-Mg)移动, 从而加强Fe3d-O2p轨道杂化和Fe—O键强度(图6). 此外, Mg主要贡献于Mg—O键中的p和s轨道, 而态密度计算显示Mg和O的轨道之间有明显重叠, 这些p和s轨道之间的成键使得轨道能量明显低于Fe3d-O2p轨道之间的杂化作用, 这有助于增强轨道间的相互作用从而提高晶格结构的稳定性. 因此, 优化后的Mg-NFS表现出优异的性能, 在0.1 C下放电比容量达到96.2 mAh/g, 并且在20 C的大倍率下仍能发挥出62.7 mAh/g的比容量. 在5 C下循环2000次后容量保持率也高达86%, 显示了优异的结构稳定性和倍率性能.
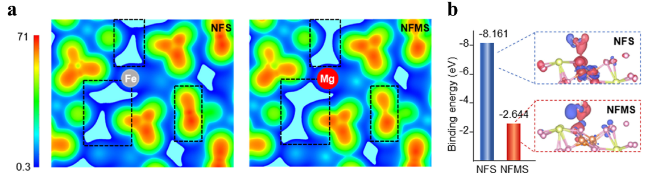
Wen等[53]注意到与Fe相比, Mg不仅表现出不同的电子态, 还具有较低的电负性(Mg: 1.2; Fe: 1.8)和较小的离子半径(Mg2+: 0.065 nm; Fe2+: 0.076 nm), 因此将Mg元素引入NFSx晶胞中取代部分Fe位点可有效调节材料(NFMS)的晶体结构和表面性质. 由于Mg较低的电负性和较小的离子半径, 掺杂后多面体结构表现出更强的键合作用和电离程度以及更小的体积, 这不仅拓宽了Na+迁移通道, 还提供了额外的路径, 使得NFMS反应动力学得到有效改善. 他们进一步研究了NFMS晶体的表面性质, 图7a展示了掺杂前后(240)晶面的表面电荷密度变化, 掺杂的Mg明显降低了材料表面电荷密度. 由于电极表面的电化学反应和相变反应大多与其表面活性电子有关, 较低的电子密度有助于抑制NFMS的表面副反应并提高其在环境中的化学稳定性. 他们采用DFT计算研究了掺杂前后材料对H2O分子的吸附能(图7b), 结果发现水分子吸附能有所降低, 证实NFMS具有更高的环境稳定性. 作者还通过CO2产气量研究了材料电极/电解液界面的稳定性. 结果显示NFMS电池产生的CO2量远低于NFS电池, 意味着更稳定的电极/电解液界面. 此外, 石英晶体微天平测试发现NFMS具有更加薄而稳定的阴极——电解质界面(CEI)膜. 因此, 归因于显著增强的Na+迁移动力学和优化的表面电荷分布, NFMS材料在10 mA/g的电流密度下显示出102.2 mAh/g的可逆容量, 并且在500 mA/g的电流密度下循环5000圈容量保持率仍高达70.8%.
对于铁基聚阴离子型正极材料, Mn、Ni是常用的有效掺杂元素[54-57]. Plewa等[58]通过简单液相法合成了Na2FeM(SO4)3 (M=Mn、Ni)材料, 并深入研究了其电化学行为. 如图8a所示, Na2FeMn(SO4)3和Na2FeNi(SO4)3的放电比容量略高于NFS, 均在110 mAh/g左右, 且电压平台随着M(即Mn和Ni)的取代而增加. 值得注意的是, DFT计算表明, 在钠脱出过程中, Na2M2(SO4)3材料中Mn3+/Mn2+和Ni3+/Ni2+氧化还原电对应存在于约4.06和5.05 V(相对于Na+/Na)处[59], 而充放电测试以及开路电压(Open circuit voltage, OCV)测试(图8a和8b)中未观察到Mn和Ni的氧化还原行为, 因此理论上Na2FeM(SO4)3 (M=Mn、Ni)材料的比容量会有一定程度下降. 但实际上, 通过进一步计算发现, 在NFS的3d晶格中引入Mn和Ni能够使化合物中的p型轨道接近费米能级, 推测Mn和Ni不直接参与电极反应, 而是类似于富锂NMC材料, 间接地将p型氧轨道推向接近费米能级, 可能触发阴离子氧的氧化还原活性, 从而提供了额外的容量[60-63].
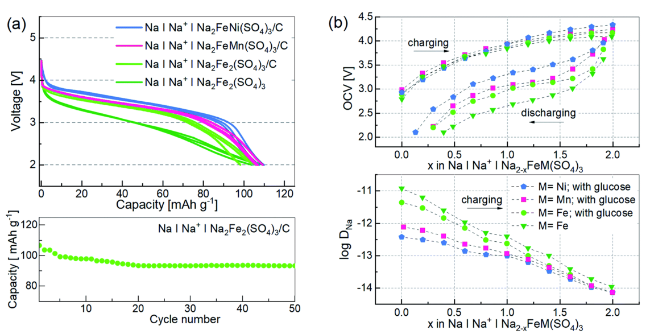
图8 (a) Na2FeM(SO4)3的前三圈的放电曲线以及NFS的循环性能; (b)充电/放电期间测量的开路电压(电流速率C/50)和第一次充电期间的钠离子扩散系数[58]Figure 8 Discharge curves for the first three cycles of Na2FeM(SO4)3 and cycling performance of NFS. (b) Open circuit voltage measured during charging/discharging (current rate C/50) and the sodium ion diffusion coefficient during the first charging[58] |
5.2.2 阴离子掺杂
SO2- 4基团丰富的电子密度是造成NFSx材料界面稳定性差的主要原因, 因此通过阴离子掺杂取代掉部分SO2- 4基团, 适当降低其高表面电子密度, 可以有效提高NFSx材料的界面稳定性.
铁基磷酸盐通常表现出良好的空气稳定性, 并且考虑到PO3- 4和SO2- 4基团半径相近, 将PO3- 4基团部分取代SO2- 4可提高NFSx材料的界面稳定性[64]. Liu等[65]采用球磨法合成了阴离子位点PO3- 4掺杂的Na2.6+xFe1.7(SO4)3-x- (PO4)x(NFSP-x)材料, 对比掺杂材料和未掺杂材料暴露在相对湿度约为50%的空气中一周前后的XRD曲线, 发现未掺杂的NFS主峰强度大大减弱, 并且出现明显的对应Na2Fe(SO4)2•4H2O的衍射峰. 相比之下, PO3- 4掺杂后的NFSP-0.3材料并未观察到明显的结构变化, 显示出增强的界面稳定性. 同时暴露后两种材料的充放电曲线也显示出截然不同的结果, NFS材料的工作电压和可逆容量显著降低, 而NFSP-0.3材料仍保持其原始的充放电特性. 为了探究PO3- 4掺杂提升NFS材料空气稳定性的机理, 采用DFT计算了NFS和NFSP-0.3材料分别对H2O分子的吸附能. 结果发现, NFSP-0.3对水的吸附能(-0.16 eV)远低于NFS材料 (-1.04 eV), 表明PO3- 4掺杂可以削弱对H2O的吸附, 从而增强了材料的界面稳定性.
优化NFSx材料的CEI膜也是一种提高其界面稳定性的重要手段. Zhang等[66]制备了F−掺杂的Na2.2Fe1.75(SO3.9F0.1)3 (NFSF)材料, 并探究了F−掺杂对材料电子状态和CEI膜性质的影响. 他们通过X射线光电子能谱(X-ray photoelectron spectroscopy, XPS)表征发现F−掺杂引起的电荷重新分布导致Fe更容易接受电子从而倾向于以Fe2+形式存在, 这激活了更多Na+参与氧化反应并增加了可逆容量. 透射电子显微镜(Transmission electron microscope, TEM)测试显示循环后的NFSF材料具有更均匀的CEI膜, 这主要是由于F−掺杂削弱了氧在正极界面的亲核性, 从而阻止电解液分解在正极界面聚集形成不均匀的CEI膜. 优化的CEI膜有利于正极界面的Na+迁移, 使得NFSF材料具有更小的电荷转移电阻, 从而在12 mA/g电流密度下初始放电比容量达到121.5 mAh/g, 在600 mA/g下循环1000圈后的容量保持率为78.8%.
5.3 碳包覆技术
碳包覆[67-69]是提升电池材料电导率的常用改性手段. 在材料外部引入一层或者多层碳包覆层不仅可以有效提升材料表面活性粒子的电导率, 还可以抑制材料颗粒的生长, 减小颗粒尺寸[70-76]. Na4Fe3(PO4)2P2O7[77-78]、Na3V2PO4[79-80]等聚阴离子型正极材料通常是通过引入有机碳源, 并经过高温碳化来生成碳包覆层. 但对于NFSx材料来说, 温度超过400 ℃时会发生分解[81], 通常煅烧温度不超过400 ℃, 显然不能达到有机碳源的碳化温度, 因此通过有机碳源高温碳化形成碳包覆层的传统方法并不适合NFSx. 采用无机导电碳材料直接作为碳包覆层可以有效避免高温碳化这一步骤, 是NFSx材料目前常用的界面改性手段. Zhu等[36]通过简单的固相合成方法分别制备了科琴黑(KB)、Super P (SP)、乙炔黑(AB)和导电石墨KS6等无机导电碳包覆的NFS材料, 并研究了碳材料种类对电化学性能的影响. 他们发现引入导电碳后, 材料的粒径有明显减小, 说明导电碳能有效避免NFS材料之间的团聚. 减小的粒径能有效缩短Na+扩散路径, 提高了Na+扩散动力学. 同时, 导电碳的引入有利于避免制备过程中Fe2+氧化成Fe3+, 并且其较大的比表面积能够促进电解液的浸润, 改善电化学性能. 其中, NFS@KB具有最大的比表面积, 表现出最高的可逆容量(85.7 mAh/g)和最佳的倍率性能(5.0 A/g下61.5 mAh/g的比容量), 并且在1000次循环后容量为74.6 mAh/g.
碳纳米管(CNT)因其优异的导电性, 也常被用作包覆材料来提高正极材料的电导率[82]. CNT覆盖在颗粒表面或缠绕在颗粒之间形成的导电网络能够显著增强材料颗粒之间的电子传输, 并且还可以抑制材料颗粒团聚, 增大活性材料与电解液的接触面积. 但是碳纳米管在水溶液的分散性差, 即使在磁力搅拌或超声分散后也只能形成悬浮液, 在使用喷雾干燥法进行合成时, 难以在水中与原料混合均匀, 导致包覆效果较差. Li等[83]提出添加十二烷基硫酸钠作为表面活性剂, 通过球磨、喷雾干燥联合制备了高容量NFSx@CNT复合材料. 十二烷基硫酸钠不会引入杂质离子, 且能有效降低碳纳米管与前驱体溶液之间的表面张力, 增强其润湿性, 使其能够在前驱体溶液中更好地分散, 显著减少碳纳米管之间的团聚现象, 最终获得碳材料在颗粒表面分布均匀的NFSx@CNT复合材料, 表现出优异的倍率性能(5 C下放电比容量为78.8 mAh/g). Yang等[84]通过喷雾干燥法合成了不同比例碳纳米管修饰的NFSx@n%CNT (n=1、2、5、10)材料, 探究了碳纳米管添加量对材料电化学性能的影响. 研究显示碳纳米管均匀覆盖在颗粒表面, 且碳纳米管的添加明显增加了材料比表面积和孔隙体积, 为钠离子的脱嵌提供了更多的活性位点, 也为电解液渗透提供了更多通道. 进一步电化学性能测试发现, 碳纳米管添加过少对材料电导率的提升效果不明显, 而添加过多则会降低材料比容量, 因此NFSx@2%CNT材料表现出最佳的电化学性能, 0.05 C下放电比容量95.9 mAh/g, 2 C下循环250圈后容量保持率高达94.7%.
与碳纳米管类似, 石墨烯(GO)的二维片层结构能够有效包裹材料颗粒, 形成覆盖整个电极的连续导电网络, 大幅降低颗粒间的接触电阻, 在提升材料电导率方面也具有显著作用[85-86]. Fang等[87]通过喷雾干燥法将Na2.4Fe1.8(SO4)3纳米颗粒均匀锚定在还原石墨烯(rGO)纳米片上(Na2.4Fe1.8(SO4)3@rGO). 纳米级颗粒有效缩短了电子/离子的扩散距离, 而石墨烯形成的3D导电网络实现了高导电基体. 该材料表现出优异的倍率性能和循环寿命(50 C下比容量为45 mAh/g, 10 C下循环10000圈后容量保持率58%). Liu等[88]同样通过喷雾干燥法合成了与rGO复合的NFSx微球材料(NFSx@rGO), 并探究了石墨烯的添加对材料形貌的影响. 他们发现未添加石墨烯的NFSx在喷雾干燥后出现一些不规则的微球, 且表面出现破碎的颗粒, 而NFSx@rGO则为规则的球形颗粒形貌. 通过机械超声处理从NFSx@rGO微球中去除NFSx纳米颗粒发现剩余的石墨烯微球呈现与NFSx@rGO相似的微球形貌, 这说明在合成过程中GO可以起到类似粘结剂的作用, 将溶液状态的反应物结合到GO表面. 同时, NFSx颗粒嵌入在石墨烯片中, 可以抑制其在煅烧过程中的生长和聚集, 有助于获得纳米颗粒. NFS@rGO微球比表面积明显降低, 这有效减小了与环境中水和氧气的接触面积, 提高了材料在空气环境中的稳定性.
与碳原子相比, 氮原子相对富含电子, 因此通过氮掺杂可以有效提高碳材料的电子导电率[89]. 此外, 氮掺杂还可以通过产生许多外在缺陷和活性位点来改变材料的结构. Wang等[90]采用低温技术合成了氮掺杂石墨烯, 并进一步通过共沉淀法制备了氮掺杂石墨烯包覆的NFSx@N-rGO材料, 通过四探针电导率测试发现, 相对于石墨烯包覆的NFSx材料(0.193 S/cm), N掺杂石墨烯能够将其电导率提高至1.173 S/cm. 此外, 氮掺杂石墨烯包覆在材料表面还能够保护内部Fe2+不被空气氧化, 增强了材料的稳定性. 得益于增强的电导率和空气稳定性, 制备的NFSx@N-rGO在0.05 C下表现出93.2 mAh/g的可逆容量, 在1 C下循环200圈容量保持率为87%.
除了上述科琴黑、碳纳米管和石墨烯等常见的导电碳材料, Di等[91]采用一种生物质碳材料——稻壳衍生炭作为复合材料提高NFSx材料的电导率. 通过酸化、高温热解等一系列过程, 可以获得具有孔径均匀、比表面积大、多孔互连等优点的稻壳炭(RHC), 进一步与原料进行球磨制备了NFSx@RHC材料, 表现出优异的倍率性能, 在5 C的高电流密度下, 比容量为60 mAh/g.
5.4 形貌/微观结构设计
通过特定形貌/微观结构设计可以有效缩短Na+扩散路径, 增强材料电导率和界面稳定性. Chen等[37]首次报道了一种具有独特设计的碳涂层和GO基体的NFS@C@GO材料, 颗粒被一层或多层GO紧密包裹, 不仅能够提供快速的电子传输, 还能有效隔绝环境中的水和氧气, 提高环境稳定性. DFT计算结果显示GO不仅能提供高电子导电介质, 还通过构建的碳网络提供了额外的电子途径, 这主要归因于NFS的O和GO的H之间形成的氢键. 同时NFS@C@GO在充放电过程中还表现出高Na+扩散系数(10−12~10−18 cm2/s)、低体积变化(<2%)和高赝电容贡献(27.9%). 因此, NFS@C@GO在0.1 C下表现出107.9 mAh/g的高比容量, 且在0.2 C下循环300圈容量保持率高达90.1%.
传统电极通常需要使用惰性的粘结剂, 不仅阻碍了活性材料之间的电子传输, 还增加了电极的重量和体积, 降低了电池的整体能量密度. 近年来, 用于钠离子电池的自支撑电极因其能够实现更高的能量密度, 提高电导率以及适用于柔性钠离子电池应用而受到了广泛关注. 自支撑电极去除了粘结剂的使用, 通常使用碳基基底制成. 这些基底用作活性材料的支架, 提供额外的电子和离子传输路径, 表现出良好的导电性[92]. Yu等[93]采用静电纺丝和静电喷涂技术制备了一种以多孔碳纤维薄膜(PCNF)为基底的NFSx@PCNF自支撑混合薄膜电极(图9a~9d). 硫酸盐纳米颗粒包裹在多孔的类石墨碳纳米纤维中, 而碳纤维相互连接构建成一个大型导电网络, 不仅保证了薄膜良好的机械性能, 其互连空隙的分层多孔结构还有利于电解液渗透和快速的钠离子迁移. 最终, 得益于高导电性、多孔框架以及纳米颗粒增强的电子/离子传输能力, NFSx@PCNF自支撑混合薄膜电极在40 C和5 C高倍率下交替循环500圈后仍能保留95%以上的初始容量.
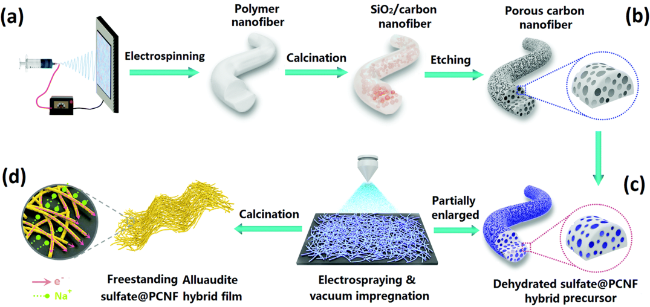
5.5 多尺度界面工程
多尺度界面工程是一种优化CEI膜性质的手段, 近年来吸引了研究者们的广泛关注. Zhang等[95]通过在Na2.26Fe1.87(SO4)3体相中引入具有密集Na+迁移通道和低迁移势垒的次要离子导体Na6Fe(SO4)4相, 构建异质结构和具有多尺度的连续Na+迁移通道. 与体相Na2.26Fe1.87(SO4)3的一维Na+迁移通道相比, Na6Fe(SO4)4不仅具有连续且广泛的三维Na+迁移通道, 并且在各方向的Na+迁移表现出更低的能量势垒, 因此它可以有效增强体相Na2.26Fe1.87(SO4)3的离子电导率和反应动力学. 通过理论计算确定了具有最低的表面电子密度的(11-2)晶面为Na2.26Fe1.87(SO4)3的可能暴露晶面, 并根据高角环形暗场扫描透射电子显微镜(HAADS-STEM)图像进一步确定了丰富的(11-2)晶面暴露在颗粒外围. 由于(11-2)晶面较低的表面电子密度, 电解液中亲核强极性FEC和ClO- 4分子在该晶面具有更强的吸附行为, 同时又不容易发生断键分解, 这会促进材料表面形成具有丰富无机物质的稳定CEI膜. 稳定的CEI膜不仅能有效抑制电解质的连续分解, 而且还赋予了材料快速的界面离子迁移动力学. 因此, 具有多尺度界面的Na2.26Fe1.87- (SO4)3/Na6Fe(SO4)4材料表现出高可逆容量(101.3 mAh/g)、优异的倍率性能(1200 mA/g下可逆容量高达73.5 mAh/g)和出色的循环稳定性(60 mA/g下循环1300圈容量保持率80.69%), 在与FeS基负极组装成全电池后能提供具有竞争力的能量密度(168.2 Wh/kg).
6 硫酸铁钠材料产业化
6.1 硫酸铁钠材料产业化挑战
硫酸铁钠作为一种成本低廉、工作电压高、循环稳定性较好的钠离子电池正极材料, 在当前锂离子电池因碳酸锂价格不断下降对钠离子电池成本要求到极致的背景下, 具有极其重要的研究和应用价值. 硫酸铁钠若成功实现规模化生产, 可以极大缓解当前市场电化学储能系统的成本压力, 提升钠离子电池的竞争力. 尽管硫酸铁钠正极在基础研究阶段取得了一定的进展(表1), 但其规模化生产仍面临着以下关键问题: (1)吸湿性使得材料储存成本增高, 阴离子掺杂[96]或界面改性[97]有望改善硫酸铁钠材料的吸湿性问题, 但仍处在实验室探索阶段; (2)合成工艺与环境要求高, 需要严格控制原料纯度以及合成过程中环境的湿度和温度; (3)杂质和低本征电导率问题使得材料容量发挥有限, 需要探索更加高效的低成本碳复合[98]以及元素掺杂[99]方案.
表1 不同设计策略的硫酸铁钠性能对比表Table 1 Performance comparison table of sodium iron sulphate with different design strategies |
| 化学式 | 改性策略 | 合成方法 | 倍率及循环性能 | 参考文献 |
|---|---|---|---|---|
| Na2+2xFe2-x(SO4)3 | 非化学计量比设计 | 喷雾干燥法 | 0.1 C, 93.8 mAh/g 20 C, 67.84 mAh/g 10 C, 1000圈, 71.1% | [41] |
| Na2.4Fe1.6Mg0.02(SO4)3 | 阳离子掺杂 | 喷雾干燥法 | 0.1 C, 96.2 mAh/g 20 C, 62.7 mAh/g 5 C, 2000圈, 86% | [52] |
| Na2.9Fe1.7(SO4)2.7(PO4)0.3 | 阴离子掺杂 | 固相球磨法 | 0.1 C, 100.4 mAh/g 30 C, 86.7 mAh/g 30 C, 6000圈, 85.5% | [65] |
| Na2.2Fe1.75(SO3.9F0.1)3 | 阴离子掺杂 | 固相球磨法 | 0.1 C, 121.5 mAh/g 5 C, 86.4 mAh/g 5 C, 1000圈, 78.8% | [66] |
| Na2+2xFe2-x(SO4)3@C@2%CNTs | 碳包覆 | 喷雾干燥法 | 0.05 C, 95.9 mAh/g 2 C, 80 mAh/g 1 C, 100圈, 96.1% | [84] |
| Na2.4Fe1.8(SO4)3@rGO | 碳包覆 | 喷雾干燥法 | 0.05 C, 100 mAh/g 50 C, 45 mAh/g 10 C, 10000圈, 58% | [87] |
| Na2+2xFe2-x(SO4)3@RHC | 碳包覆 | 固相球磨法 | 0.1 C, 96.8 mAh/g 20 C, 30 mAh/g 0.1 C, 100圈, 83.9% | [91] |
| Na2Fe2(SO4)3@C@GO | 微观结构设计 | 冷冻干燥法 | 0.1 C, 107.9 mAh/g 10 C, 75.1 mAh/g 5 C, 800圈, 80.1% | [37] |
| Na2+2xFe2-x(SO4)3@SWNT | 微观结构设计 | 静电纺丝法 | 0.05 C, 90.8 mAh/g 10 C, 74.9 mAh/g 5 C, 100圈, 92% | [94] |
| Na2.26Fe1.87(SO4)3/Na6Fe(SO4)4 | 多尺度界面工程 | 冷冻干燥法 | 6 mA/g, 101.3 mAh/g 1200 mA/g, 73.5 mAh/g 60 mA/g, 1300圈, 80.69% | [95] |
6.2 硫酸铁钠材料产业化现状
目前学术界关于聚阴离子储钠正极材料的研究仍以磷酸盐为主, 硫酸盐因为制备难度大和使用相对困难, 研究和应用的门槛较高, 因此基础研究相对偏少. 中南大学, 武汉大学, 苏州大学, 郑州大学, 复旦大学等国内高校的相关课题组已开展了较多的工作. 部分企业均依托相关高校的研究团队的研究成果, 进行产业化推进. 近年来, 国内主流高校和企业积极布局硫酸铁钠材料专利, 截至2025年6月, 关于硫酸铁钠材料专利, 各企业累计申请128项, 高校累计申请21项. 表2分析了一些代表性企业对硫酸铁钠材料的具体专利布局情况. 从表2可见, 三一红象、江苏众钠、格林美能源材料、深圳先进研究院等公司和研究院所积极布局硫酸铁钠改性以及规模生产相关方面的专利, 致力于推动硫酸铁钠材料的商业化应用. 众钠能源自2021年开始便深耕于硫酸铁钠材料技术和电池技术开发, 已在轻型动力电池、启动电源、工商业储能等领域推出了多元场景解决方案. 据其公司网页介绍, 已规划启动建设1万吨硫酸铁钠的量产线. 针对轻型动力市场, 众钠能源推出的25 Ah硫酸铁钠电池产品循环寿命超过2000次, 并在 -20~50 ℃的宽温域下能够正常工作. 华钠新材也积极参与硫酸铁钠产业化布局, 目前建设有千吨级聚阴离子硫酸铁钠的产线, 其产品可应用于微型车、电动工具以及家庭储能等场景. 其宣称在2027年硫酸铁钠体系的钠离子电池成本有望降低至0.2元/Wh. 华友新材料也自2022年初着手布局钠电产业, 并明确“坚定聚阴离子硫酸铁钠路线研发”方向, 围绕资源战略, 快速搭建研发及制造平台, 已成功实现从产品中试、大试工艺定型到3600吨/年量产产线建设投运, 初步形成了一条具有竞争力的低成本、短流程、绿色工艺路线, 并在2024年实现了钠电聚阴离子硫酸铁钠前驱体量产并顺利交付. 上海派能科技公司推出的硫酸铁钠24 Ah软包电池产品常温循环寿命可达2000次, 可满足轻型电动车出行需求. 英钠能源、珈钠能源等企业也在积极攻关硫酸铁钠材料, 致力于突破其结构稳定性差、吸水性强、高温性能差等问题. 根据高工产业研究院(GGII)预计2030年钠离子电池聚阴离子正极材料市场规模或将达到120 GWh, 而硫酸铁钠有望占据其中部分份额, 在储能、启停电源、两轮车等领域实现规模化应用.
表2 硫酸铁钠材料代表性专利申请统计表Table 2 Patent landscape analysis of sodium iron sulfate materials |
| 申请公司 | 发明名称 | 针对问题 | 创新点 | 参考专利 |
|---|---|---|---|---|
| 三一红象电池有限公司 | 一种碳复合硫酸铁钠正极材料及其制备方法和应用 | 压实密度低 | 混料方式: 砂磨 | [98] |
| 一种制备碳包覆硫酸铁钠材料的方法、碳包覆硫酸铁钠材料和电池 | 无机碳源包覆不均匀 | 引入分散剂: 十六烷基三甲基溴化铵 | [96] | |
| 一种制备碳包覆硫酸铁钠材料的方法、碳包覆硫酸铁钠材料和电池 | 无机碳源包覆不均匀 | 球磨预分散无机碳源 | [100] | |
| 深圳先进研究院 | 一种硫酸铁钠复合材料及其制备方法与应用 | 比容量偏低 | 与类普鲁士蓝材料复合 | [101] |
| 一种双掺杂硫酸铁钠正极材料及其制备方法和应用 | 本征电导率低 | 阴阳离子双掺杂 | [102] | |
| 一种硫酸铁钠材料及其制备方法和应用 | 空气稳定性差 | 非晶硫酸铁钠表面包覆 | [103] | |
| 一种硫酸铁钠及其制备方法和应用 | 振实密度 | 微波溶剂热反应 | [104] | |
| 一种自支撑正极及其制备方法和应用 | 本征电导率低 | 自支撑电极设计 | [105] | |
| 深圳市津工能源有限公司 | 一种碳包覆钾锆共掺杂的硫酸铁钠正极材料及其制备方法和应用 | 本征电导率低 | 多阳离子共掺杂 | [106] |
| 江苏众钠能源科技有限公司 | 一种复合正极材料、正极片及二次电池 | 比容量偏低、高温循环稳定性差 | 碳包覆磷酸焦磷酸铁钠材料包覆 | [107] |
| 一种复合钠离子正极材料、制备方法及钠离子电池 | 易生成杂质、本征电导率低 | 分层式结构设计 | [108] | |
| 一种碳包裹的硫酸铁钠正极材料及其制备方法 | 本征电导率低 | 氮化碳基材料包覆 | [109] | |
| 超威电源集团有限公司 | 一种表面改性的硫酸铁钠正极材料及其制备方法 | 空气稳定性差 | 疏水材料表面改性 | [97] |
| 贵州为方能源新材料科技有限公司 | 碳包覆掺杂铕的硫酸铁钠正极材料及其制备方法、钠离子电池和用电设备 | 本征电导率低 | 阳离子掺杂 | [110] |
| 格林美(无锡)能源材料有限公司 | 一种硫酸铁钠正极材料及其制备方法 | 本征电导率低 | 阳离子掺杂 | [111] |
7 结论与展望
锂离子电池作为成熟的电化学储能体系, 占据了市场的大部分份额. 但锂资源不足和安全性等问题使锂离子电池仍面临严峻挑战. 具有钠资源丰富, 成本低廉, 安全性更好的钠离子电池得到了科研工作者的广泛关注. 正极材料是决定钠离子电池成本最主要的部分, 探索高性能低成本的正极材料是快速推进SIBs应用的关键. 硫酸铁钠材料具有工作电压高、结构稳定、成本低廉、环境友好等优点, 被视为钠离子电池极具商业化价值的正极材料之一.
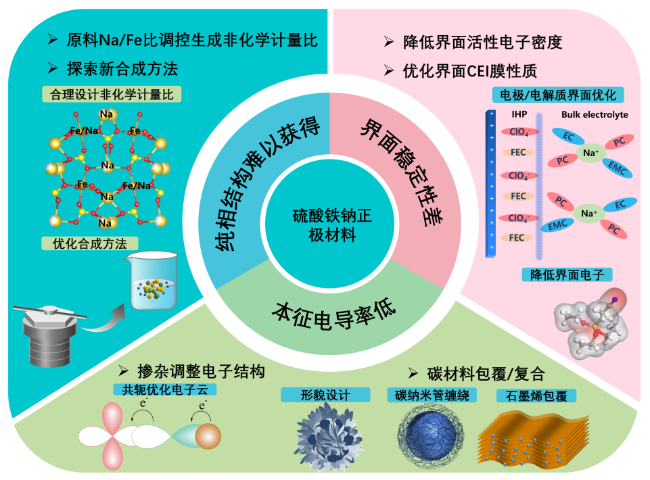
本综述介绍了硫酸铁钠材料的晶体结构及钠离子脱嵌机制, 总结了所面临的关键问题, 提出了系列提升策略, 并概述了其产业化进展. NFS材料具有稳定的Alluaudite型三维框架, 为钠离子的快速脱嵌提供了结构保障, 且3.8 V的高工作电压保证了较高的能量密度. 但如图11所示, NFS材料仍然面临着以下关键问题: (1)纯相结构难以获得, 影响电化学性能的发挥. Fe2O10二聚体中Fe2+强烈的库仑斥力使得NFS材料中Na占据Fe位点从而趋向于生成更稳定非化学计量比NFSx材料, 因此合理调控原料中Na/Fe比有利于减少杂质含量, 同时有必要探索新的合成方法减少杂质含量; (2)本征电导率低. 掺杂和碳材料包覆复合是有效提高材料电导率的手段, 探索更加丰富的掺杂元素以及构建薄而有效的碳层是未来进一步提高NFS材料电导率的方向; (3)界面稳定性较差. 具有强烈诱导效应的SO2- 4基团为NFS材料提供了高工作电压, 但也给其带来了丰富的表面电子密度, 降低了界面稳定性. 通过掺杂调控其表面电子密度以及优化电极/电解质界面CEI膜性质是提高其界面稳定性的有效手段.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
为了进一步满足高性能钠离子电池对低成本高比能长寿命正极材料的需求, 硫酸铁钠材料开发还需要更多基础性的研究, 也需要更多研究者们的关注. 本综述在前面讨论的基础上, 对硫酸铁钠材料的下一步研究提出以下展望: (1)基于NFS材料的高工作电压, 通常需要充电至4.5 V左右的高压, 常用的电解液在此高压下容易发生分解, 造成界面的不稳定. 因此, 亟需开发匹配的高压电解液来提升硫酸铁钠材料稳定性和能量密度. (2)目前对NFS材料的改性策略比较单一, 性能提升有限. 可以借鉴其他正极材料改性手段, 如构建异质结构、表面涂层以及熵焓调控等策略. (3)对于NFS材料暴露在空气中的失效机制研究较为浅显, 有必要借助先进表征技术和理论计算来探究NFS材料在空气中失效的深层次机理, 从而为设计出具有良好的空气稳定性的NFS材料奠定理论基础. (4)总体来说, NFS材料的研究仍处于起步阶段, 对于其后续的产业化仍需要基础研究的重要支撑, 同时需要各企业积极开发低成本、高性能NFS材料的绿色规模产线, 从而推动其商业化进程.
(Cheng, B.)