

1 引言
近些年来, 钙钛矿型复合氧化物由于其成本较低、合成难度低、热稳定性高, 在脱除NOx和CO方面是最有前途的催化剂, 受到了国内外学者的广泛研究[7-10]. 其化学式为ABO3, A位离子起到稳定结构的作用[11], 一般为+3价稀土或+2价碱土金属离子, 位于立方体的中心, 与12个氧配位; B位一般为+3价或+4价过渡金属离子, 位于八面体的中心, 与6个氧配位, B位离子的种类及价态是决定催化活性的主要因素, 当价态变化时会形成晶格缺陷和大量氧空位, 有利于晶格氧的迁移和扩散以及表面吸附氧增多. 研究表明, 在满足容限因子的条件下, 通过A位或B位掺杂可改变元素化合态及氧化还原性能, 会产生更多晶格缺陷和氧空位以及更多的酸性位点, 氧空位有利于NO和O2物种的活化, 使催化剂具有更高效的催化活性[11-14].
在往年的研究中, 学者通过A位引入Sr2+改变了钙钛矿B位阳离子的还原性, 改变了Mn4+/Mn3+的比值, 增加了Oads/Olatt比值, 产生了结构缺陷, 使氧更容易扩散, 从而提高催化活性. 例如Guo等[15]研究发现La1–xSrxMnO3用于降解水中的RhB, 降解速率明显增强可以到达96%以上的降解率. Pei等[16]采用溶胶-凝胶法制备了La0.85Sr0.15MnO3催化剂, 样品在反应中展现出最佳活性, 在50 ℃下反应3 h, BzOH转化率达到97.0%. 当Fe掺杂到钙钛矿的B位时, 它们会改变B位金属阳离子的价态, 并有效提高氧化物表面的晶格缺陷及氧空位数量, 进一步增强了催化活性. Wu等[17]研究发现, 替代部分Mn离子的过渡金属离子, 如Cu、Co、Fe、Ni、Cr等, 都能提高催化剂的脱硝效率. 同时, 根据最近研究已经发现将金属离子共掺杂到位点A和B可以进一步提高其催化活性并确保其结构稳定性. Tarjomannejad 等[18]采用溶胶-凝胶法合成了LaMn1–xFexO3 (x=0, 0.3, 0.5, 0.7, 1)和La0.8A0.2Fe0.7Mn0.3O3 (A=Sr, Cs, Ba, Ce)钙钛矿型氧化物, Sr2+在钙钛矿A位的引入改变了B位阳离子的还原性, 改变了Fe4+/Fe3+和Mn4+/Mn3+的比值, 增加了Oads/Olatt比值, 产生了结构缺陷, 导致了较高的催化活性. Liu等[19]通过A、B位协同调控制备的La0.8Ce0.2Mn0.6Fe0.4O3, 催化性能的改善和催化剂表面Mn4+-O-Mn3+和Mn4+-O-Fe3+活性物种的高度分散密切相关, 适量Fe掺杂有利于CO吸附和活化, CO捕获表面晶格氧生成CO2, 导致氧空位(SOV)和界面氧空位(SSOV)的形成, NO可以吸附在这些氧空位上进行解离, 生成N2O或N2, 展现出优异的CO-SCR性能.
尽管目前的研究已经明确了A/B位掺杂能够增强LaMnO3的催化活性, 并且有学者基于密度泛函理论(DFT)阐明了未掺杂LaMnO3催化剂表面的CO-SCR反应机理, 但掺杂改性的同时可能伴随着反应机理的变化. 对于Sr/Fe掺杂LaMnO3的CO-SCR催化机理、反应路径及抗毒性能的影响, 目前尚不明确, 包括表面气体的吸附及活化行为、晶格氧的迁移机制以及N2的生成路径等. 在微观层面对上述机制进行明确阐释具有重要意义, 将为设计兼具低温高效和抗中毒能力强的LaMnO3脱硝催化剂提供理论指导.
因此, 本文基于密度泛函理论系统探究了LaMnO3掺杂体系的表面反应机理, 通过对比分析掺杂体系的表面吸附能、几何构型及电子结构特征, 深入解析Sr/Fe掺杂LaMnO3对催化剂表面的吸附行为与反应机理的影响. 其中重点探索了CO在LaSrMnFeO3催化剂表面的氧化反应路径和选择性催化还原(SCR)机理.
2 结果与讨论
2.1 Sr/Fe掺杂体系计算模型的构建
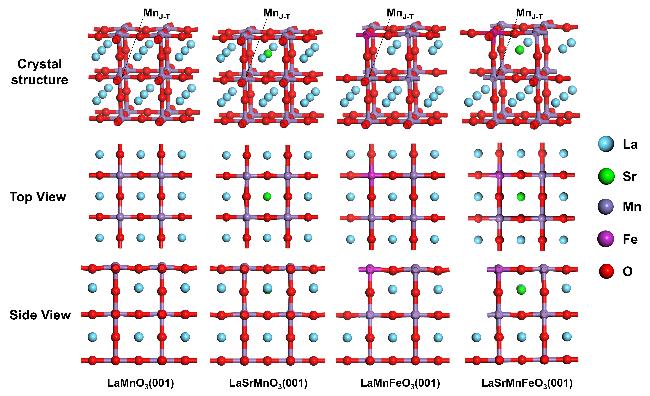
在LaMnO3以(001)断面截断时, 其原子的配位环境发生变化, 形成了两种离子性质不同的终端面, 分别为MnO2终端面和LaO终端面, 为了确定哪个端面有利于CO/NO的吸附, 对其进行了弛豫计算. 结果表明: MnO2终端面较LaO终端面有着更低的表面能, 说明MnO2终端面比LaO终端面更为稳定, 这可能是由于La原子相较于Mn原子失去了更多的O近邻. 且MnO2终端面更容易暴露于大气中. 因此, 在考虑计算效率和精度的基础上, 本文构建了含有5层原子的LaMnO3(001)模型, 选择MnO2为终端面进行模拟研究. 在计算过程中, 允许上部两层原子自由弛豫, 其中最底部的3层原子保持固定, 以防止相邻层之间的相互作用. 并在其上方设置1.5 nm的真空层. 采用了对称平板构型进行计算, 消除偶极矩的影响. 此外, 本文根据组内实验La1–xSrx-Mn1–yFeyO3系列不同配比钙钛矿中, Sr和Fe掺杂比例均为0.1时在CO-SCR的脱硝效率最高且N2选择性高. 基于此, 对LaMnO3(001)掺杂体系依据0.1比例进行了模型的构建, 并对此进行了结构优化计算. 如图1所示.
2.2 Sr/Fe掺杂对LaMnO3结构的影响
LaMnO3掺杂体系弛豫计算结果表明: A位掺杂Sr后的模型膨胀最为明显, 说明八面体发生了形变. 相较于未掺杂的LaMnO3, A、B位掺杂使得Mn—O键长在x、y轴方向有一定程度拉伸, z轴方向被压缩, 第一层与第二层之间的间距膨胀, 第二层与第三层压缩, 同时 Mn—O—Mn键角在3个方向也逐渐减小, 八面体发生压缩畸变, 这是因为Mn d轨道分裂的eg简并电子在dz2轨道上, 畸变之后dx2-y2上升, dz2下降, 简并消除, 对x, y轴上斥力减小, 所以轴向键伸长而赤道键缩短, 形成不同键长的Mn—O键, 导致晶格畸变加剧, 可能有利于形成氧空位. 一方面, Sr2+原子半径(0.112 nm)比La3+(0.106 nm)更大, 会直接导致晶格畸变; 另一方面, 当低价态Sr2+取代LaMnO3晶格中La3+时, 会产生净电荷失衡, 需要通过电荷补偿机制来维持电中性, 部分金属离子的氧化态升高, 或通过产生氧空位实现电荷补偿, Mn3+倾向于变为Mn4+, 造成晶格收缩, 晶胞体积减小, 而LaMnO3的比表面积会增加, 这提供了更多暴露的活性位点, 有利于后续分子的吸附. 与此同时, 部分Mn—O键因离子半径变小而略微拉长, 这也使得Mn在晶格中具有更高的反应性[23-24]. 此外, 因为Fe2+ (0.076 nm)、Fe3+ (0.064 nm)或Fe4+ (0.059 nm)的离子半径小于Mn2+ (0.08 nm)、Mn3+ (0.066 nm)、Mn4+ (0.054 nm)离子半径, 半径越小键长越短, 掺杂Fe时, 导致Fe—O键伸长, 晶体为维持结构稳定性, 与之相邻的第二层Mn—O键因晶格应力略微压缩, 同时Q的值也随着掺杂增大, 大于初始LaMnO3的–0.007 nm. 这说明掺杂后存在较强的Jahn-Teller效应, 而其中Sr-Fe共掺杂产生协同效应, 使MnO6八面体畸变最为显著, Q值到达 –0.0166 nm.
2.3 Sr/Fe掺杂对CO吸附性能的影响
首先在CO-SCR反应过程中, CO作为还原剂参与NO的CO-SCR反应, 其在催化剂表面的吸附性能对后续反应过程产生重要影响. 因此, 研究了未掺杂、A位掺杂、B位掺杂、AB位共掺杂LaMnO3体系表面对CO的吸附机理. 催化剂表面的Mn和掺杂的Fe原子可以作为Lewis酸位点. CO的C端更容易垂直吸附或轻微的倾斜吸附到LaMnO3掺杂体系中的Lewis酸位上[25]. 优化后的稳定吸附构型如图2所示. 相应的吸附能、键长如表1所示. 同时考虑到LaMnO3为表面对称模型, LaMnO3(001)和LaSrMneO3(001)构型表面活性位点吸附性质一致, 在LaMnO3(001)和LaSrMnO3(001)表面分别得到了一种稳定的吸附构型, Mn01作为其表面的活性位点, 吸附能分别为–0.35、–0.44 eV. 对于LaMnFeO3(001)吸附构型, 催化剂表面的活性位点分别为Fe、Mnl、Mn2、Mn3, 其对应的吸附能分别为–0.85、–0.43、–0.43和–0.30 eV. 在LaSrMnFeO3(001)表面上得到了4种稳定的构型, 分别以CO分子的C端去靠近Fe、Mnl、Mn2、Mn3四个活性位点, 相应的吸附能分别为–0.63、–0.49、–0.41和–0.31 eV. 值得注意的是, 掺杂Fe的吸附构型, Mn3位点的吸附能明显低于Fe、Mn2、Mn1, 这因为Fe掺杂加快了电子转移并提升了其氧化还原性能(Mn3++Fe3+→Fe2++Mn4+), 增强临近Mn位点吸附活性. Mn3位点距Fe位点较远, 受影响较小.
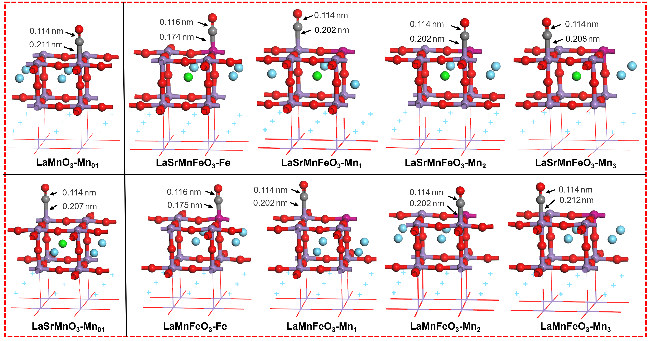
图2 CO在LaMnO3掺杂体系表面的吸附构型Figure 2 Adsorption configuration of CO on the surface of LaMnO3 doped system |
表1 CO在LaMnO3掺杂体系表面的吸附能和键长Table 1 Adsorption energy and bond length of CO on the surface of LaMnO3 doped system |
| dC-O/nm | dC-metal/nm | Eads/eV | |
|---|---|---|---|
| LaMnO3-CO-Mn01 | 0.114 | 0.211 | –0.35 |
| LaSrMnO3-CO-Mn01 | 0.114 | 0.207 | –0.44 |
| LaMnFeO3-CO-Mn1 | 0.114 | 0.202 | –0.43 |
| LaMnFeO3-CO-Mn2 | 0.114 | 0.202 | –0.43 |
| LaMnFeO3-CO-Mn3 | 0.114 | 0.212 | –0.30 |
| LaMnFeO3-CO-Fe | 0.116 | 0.175 | –0.85 |
| LaSrMnFeO3-CO-Mn1 | 0.114 | 0.202 | –0.49 |
| LaSrMnFeO3-CO-Mn2 | 0.114 | 0.202 | –0.41 |
| LaSrMnFeO3-CO-Mn3 | 0.114 | 0.208 | –0.31 |
| LaSrMnFeO3-CO-Fe | 0.116 | 0.174 | –0.63 |
计算结果表明: CO分子在Fe位点的吸附稳定性最高, 更倾向于优先占据该活性位点. 在LaMnFeO3(001)和LaSrMnFeO3(001)构型, CO吸附的最稳定构型中分别形成键长为0.175 nm的Fe—C和0.174 nm Fe—C键, 其键长明显短于Mn—C键长, Fe位点与吸附气体分子之间的键长明显收缩, 说明Fe与C端形成了更强的化学键, 且吸附在Fe位CO分子的C—O键长由初始的0.114 nm伸长至0.116 nm, 预示着下一步CO分子的解离. 表明Fe原子的掺杂提高了LaMnO3催化活性.
对比不同的掺杂体系发现, Sr单掺杂仅使吸附能从–0.35 eV微弱提升至–0.44 eV, 吸附稳定性提升有限, 难以实现稳定的化学吸附, 而Fe单掺杂使最佳吸附能达到–0.85 eV, 证实了Fe对其电子结构的调控起主导作用[26]. 此外, Sr-Fe共掺杂虽然提升了CO在催化剂表面的吸附性能, 但相较于单掺杂的Fe构型吸附能力有所减弱, 这主要是归因于Sr电子供体特性抑制Fe/Mn间的氧化还原协调效应. 为了保证后续分析的可比性, 对于LaMnFeO3(001)和LaSrMnFeO3(001)两种掺杂构型, 我们选择CO在Fe位点的最稳定吸附构型进行后续分析.
Bader电荷分析结果(表2)显示: 在LaMnO3和LaSrMnO3吸附构型的bader电荷为负值, 表明CO分子吸引了吸附底物上的孤对电子, 电子由CO分子向催化剂表面转移, 这导致吸附能力表现为不稳定吸附, 且与前文吸附能分析结果一致. 在LaMnFeO3和LaSrMnFeO3吸附构型的bader电荷为正值, 说明在吸附过程中电子由CO分子向催化剂表面转移——这是由于CO分子中存在孤对电子, 而催化剂表面存在3d空轨道足以容纳过量电子, 有利于下一步化学键的形成. 此外, 可以观察到掺杂Fe原子的吸附构型的CO分子整体的电子转移较大, 这与其在催化剂表面Fe位点的吸附能较大相一致. 值得注意的是, 掺杂Sr的构型抑制了催化剂与CO分子bader的转移, 这一现象与上文分析中Sr带来的电子调控效应表现一致.
表2 LaMnO3掺杂体系最佳CO吸附构型的Bader电荷Table 2 Bader charge for optimal CO adsorption configuration of LaMnO3 doping system |
| Q(Sr)/e | Q(Fe)/e | Q(C)/e | Q(O)/e | Q(Mn/Fe)/e | ΔQCO/e | |
|---|---|---|---|---|---|---|
| LaMnO3-CO | — | — | –0.292 | 0.246 | –1.659 | –0.046 |
| LaSrMnO3-CO | –1.659 | — | –0.061 | 0.022 | –1.710 | –0.039 |
| LaMnFeO3-CO | — | –1.364 | –0.120 | 0.238 | –1.544 | 0.118 |
| LaSrMnFeO3-CO | –1.660 | –1.436 | –0.126 | 0.236 | –1.608 | 0.110 |
图3展示了LaMnO3掺杂体系催化剂上吸附CO最稳定构型的差分电荷密度分布(CDD). 差分电荷密度分析表明, 对于LaMnO3和LaSrMnO3的CO吸附构型, 黄色区域集中在C—Mn键之间, C—Mn键积累的电子越多, 蓝色区域集中在O原子附近, O原子附近消耗的电子越多, 对比以上两种构型, LaSrMnO3吸附构型C—Mn电子积累和C原子附近的电子消耗较未掺杂催化剂较多, 表明Sr掺杂引发的晶格畸变增强了活性位点Mn的电子供给能力. 对于LaMnFeO3和LaSrMnFeO3的CO吸附构型, CO稳定吸附在Fe位点, 黄色区域集中在 C—Fe键之间, 表明C—Fe键存在电子积累, 蓝色区域集中在C—O键之间, 表明C—O键存在电子消耗, 其中LaMnFeO3吸附构型, C—Fe键和C—O之间的电子积累和电子消耗较其他吸附构型表现为消耗更多和更快. 对比四种吸附构型, C-metal键的电子积累能力可以排序: LaMnFeO3>LaSrMnFeO3>LaSrMnO3>LaMnO3, 这是归因Fe的掺杂通过引入未占据的3d轨道, 增强了其氧化还原能力, 显著提升表面的电荷密度. 上述分析进一步表明, CO分子与掺杂体系催化剂表面存在较强的相互作用, 这与前文Sr和Fe掺杂导致的吸附能变化结果相一致.
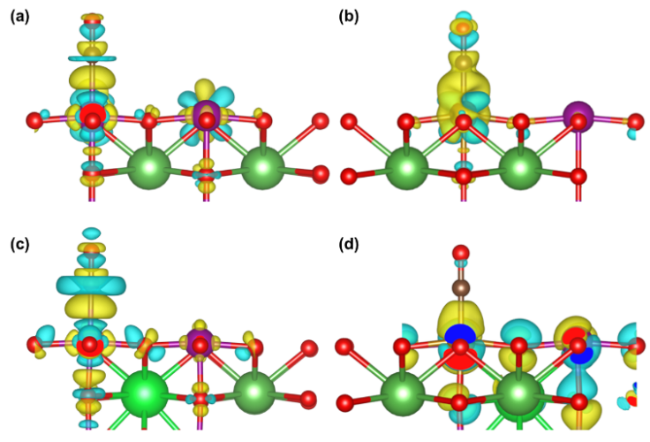
此外, 从局域电荷密度函数(ELF, 图4)可以看出, LaMnO3掺杂体系中CO吸附构型的C原子与邻近Mn或Fe原子之间形成ELF>0.8的红色电子云, 显示出高度电子局域化, 表明Mn—C和Fe—C键具有较强的共价特性.
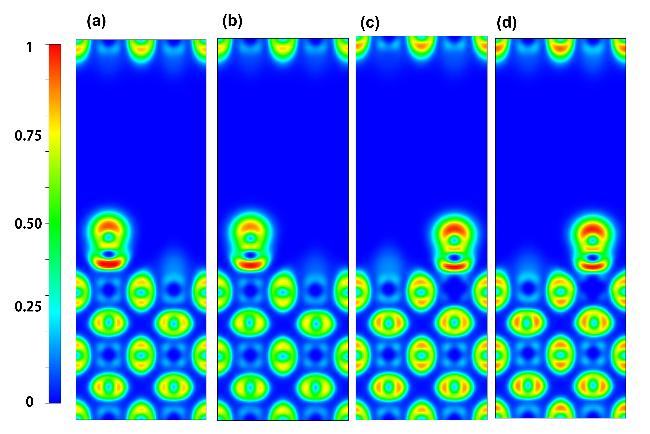
为了进一步阐明CO分子在LaMnO3掺杂体系表面的吸附机理, 对最佳CO吸附构型的投影态密度进行了详细分析. 图5展示了CO在不同LaMnO3掺杂体系表面的投影态密度分布. 在未掺杂的LaMnO3构型中, CO分子中C原子的p轨道与Mn 3d轨道在约–7.2 eV和2.3 eV处出现共振峰, 表明二者存在杂化并可能形成共价键. 然而, 2.3 eV处Mn 3d轨道峰值较高, 导致与C 2p轨道的杂化程度较弱. 在LaSrMnO3构型中, C的p轨道与Mn 3d轨道在–2.4 eV处出现共振峰, 显示出更优的轨道重叠. 在LaMnFeO3构型中, C的p轨道与Fe的3d轨道在约–6.8 eV和–2.7 eV处出现多处明显共振峰, 表明杂化效果显著, 结合相互作用增强. 同样, 在LaSrMnFeO3构型中, C 2p轨道与Fe 3d轨道在–2.5 eV处也表现出明显的共振峰. Sr、Fe及Sr-Fe共掺杂使得C 2p轨道峰值能级整体降低, 更接近Mn或Fe的3d轨道能级, 从而促进轨道间的杂化, 增强了CO的吸附和活化能力. 此外, 在掺杂了Sr的构型中, C 2p轨道在–5 eV至–7.5 eV范围内的峰值显著减弱甚至几乎消失, 这主要归因于Sr掺杂引起的电荷补偿效应, 使催化剂表面的电子态密度重新分布至更稳定的低能区域.
值得注意的是, 相较于未掺杂的LaMnO3体系, 掺杂体系中C 2p轨道峰值普遍向下移动, 且移动幅度依次为LaMnFeO3>LaSrMnFeO3>LaSrMnO3. 这主要是因为Fe的电负性高于Mn, 其未占据的3d轨道能级更低, CO的2π*反键轨道容易反馈电子, 形成强反馈π键, 显著增强CO吸附稳定性.
对于LaSrMnFeO3体系, Sr掺杂引入的电荷补偿效应与Fe掺杂产生的极化效应相互作用, 形成动态平衡, 在一定程度上抵消了单一Fe掺杂带来的过度极化. 同时, C 2p轨道在0至5 eV范围内峰强度减弱, 表明电子密度有所流失, 这与前文的Bader电荷分析结果相符.
2.4 Sr/Fe掺杂对NO吸附性能的影响
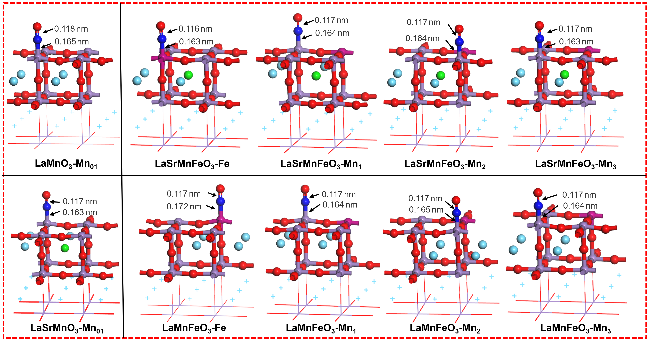
图6 NO在LaMnO3掺杂体系表面的吸附构型Figure 6 Adsorption configuration of NO on the surface of LaMnO3 doped system (a) LaMnO3, (b) LaSrMnO3, (c) LaMnFeO3, (d) LaSrMnFeO3. |
表3 NO在LaMnO3掺杂体系表面的吸附能和键长Table 3 Adsorption energy and bond length of NO on the surface of LaMnO3 doped system |
| dN-O/nm | dN-metal/nm | Eads/eV | |
|---|---|---|---|
| LaMnO3-NO-Mn01 | 0.118 | 0.165 | –1.88 |
| LaSrMnO3-NO-Mn01 | 0.117 | 0.163 | –1.76 |
| LaMnFeO3-NO-Mn1 | 0.117 | 0.164 | –1.93 |
| LaMnFeO3-NO-Mn2 | 0.117 | 0.165 | –1.93 |
| LaMnFeO3-NO-Mn3 | 0.117 | 0.164 | –1.83 |
| LaMnFeO3-NO-Fe | 0.117 | 0.172 | –1.85 |
| LaSrMnFeO3-NO-Mn1 | 0.117 | 0.164 | –1.79 |
| LaSrMnFeO3-NO-Mn2 | 0.117 | 0.164 | –1.78 |
| LaSrMnFeO3-NO-Mn3 | 0.117 | 0.163 | –1.74 |
| LaSrMnFeO3-NO-Fe | 0.116 | 0.163 | –1.54 |
Eads值如表3所示, 计算结果表明: 对于LaMnO3掺杂构型表面NO的吸附, 最终优化的结构NO原子均有效吸附在表面, 所有的NO吸附都是高度放热的, 其吸附范围从–1.54 eV到–1.93 eV. 通常, 与吸附能低于–0.31 eV的相互作用被归类为物理吸附, 而与吸附能高于–0.52 eV的相互作用被归类为化学吸附[27], 与基底发生较强的相互作用. 程序升温脱附(TPD)实验结果表明, NO解吸峰与化学吸附[28]有关. 上述计算结果与实验观察结果吻合良好. 其中, NO更倾向于吸附到Mn位点上, 在掺杂Fe的吸附构型中, Mn1位点表现出最大的吸附能. 此外, 表3中数据表明Fe单掺杂构型在掺杂体系中对NO分子的吸附能力是整体来说表现最强的, 表明Fe的掺杂能够显著增强NO在催化剂表面的吸附能力. Sr单掺杂构型对NO的吸附能绝对值明显小于未掺杂构型, Sr-Fe共掺杂构型的吸附能绝对值也明显小于单掺杂Fe构型, 表明Sr掺杂削弱了NO的吸附能力. 这可能是Sr2+取代La3+导致Mn3+氧化为高价态 Mn4+, Mn4+的d³电子构型使其电子供给能力减弱, 难以向NO的2π*反键轨道提供电子, 削弱了Mn—N化学键强度. 此外, Sr/Fe掺杂构型和未掺杂构型的NO吸附能绝对值显著高于CO吸附能绝对值, 这有助于避免NO和CO之间的竞争吸附.
NO吸附在LaMnO3掺杂体系表面的差分电荷密度如图7所示. 图中蓝色区域主要分布于N—O键之间, 表示电荷消耗; 黄色区域集中于Mn—N键之间, 表示电荷积聚, 这表明NO的部分轨道电荷发生了转移. 进一步表明NO分子与基底相应的吸附位点有很强的相互作用. 同时, 在NO和催化剂表面之间观察到相当大的互极化. 值得注意的是, 与未掺杂构型相比, Fe单掺杂构型中Mn—N键间电荷积聚更多, N—O键间电荷消耗更明显, 表明NO与催化剂表面的吸附作用更强, 这与吸附能增强的结果一致. 此外, Sr单掺杂导致Mn—N键间电荷积聚的对称性明显降低, 与之前对Sr掺杂结构影响的分析相符. 为进一步分析电荷转移行为, 计算了局域电荷密度(ELF)以验证化学键性质, 并通过Bader电荷分析定量考察电荷转移量.
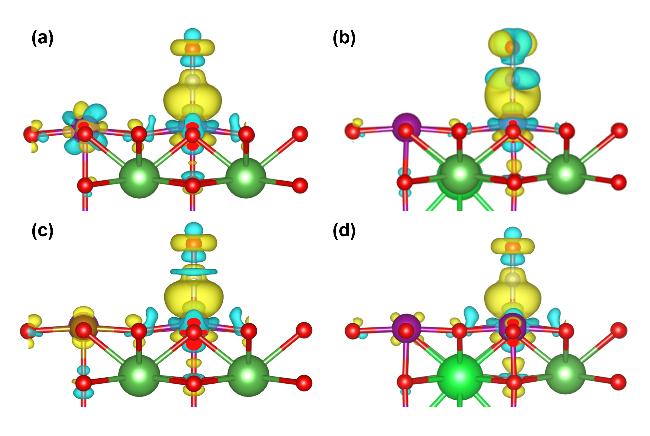
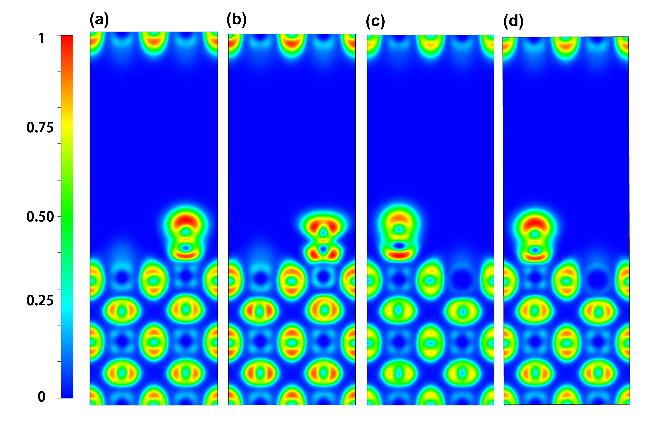
LaMnO3掺杂体系的局域电荷密度(ELF)如图8所示, 掺杂Sr和Fe都会导致电荷在NO分子中聚集, 且N原子和O原子中间键呈现黄色电子云, N原子和Mn原子之间电子云呈现黄色和红色, N原子和O原子之间电子云呈现黄色, ELF>0.8, 电子局域性高, N—Mn、N—O均表现为共价键, 其中ELFN-Mn>ELFN-O, 表明N—Mn键强度更高.
此时, 通过表4的Bader电荷定量分析发现, NO向LaMnO3掺杂体系催化剂表面输送电子, 其中主要是NO的5δ轨道向金属d轨道提供电子. 此时, 未掺杂构型中转移了0.245e电子, Fe单掺杂构型转移了0.256e电子, Sr单掺杂构型中转移了0.226e电子, Sr-Fe共掺杂构型中NO获得0.249e电子, 上述结果表明Fe的掺杂显著增强了NO与催化剂表面电子转移能力, 而Sr的掺杂减弱了NO与催化剂表面电子转移能力, 这与差分电荷密度中Mn—N键电荷积聚增加及吸附能提升的趋势一致.
表4 LaMnO3掺杂体系最佳NO吸附构型的Bader电荷Table 4 Bader charge for optimal NO adsorption configuration of LaMnO3 doping system |
| Q(Sr)/e | Q(Fe)/e | Q(N)/e | Q(O)/e | Q(Mn)/e | ΔQNO/e | |
|---|---|---|---|---|---|---|
| LaMnO3-NO | — | — | 0.112 | 0.135 | –1.636 | 0.245 |
| LaSrMnO3-NO | –1.664 | — | 0.106 | 0.126 | –1.634 | 0.226 |
| LaMnFeO3-NO | — | –1.601 | 0.110 | 0.146 | –1.641 | 0.256 |
| LaSrMnFeO3-NO | –1.658 | –1.591 | 0.148 | 0.101 | –1.698 | 0.249 |
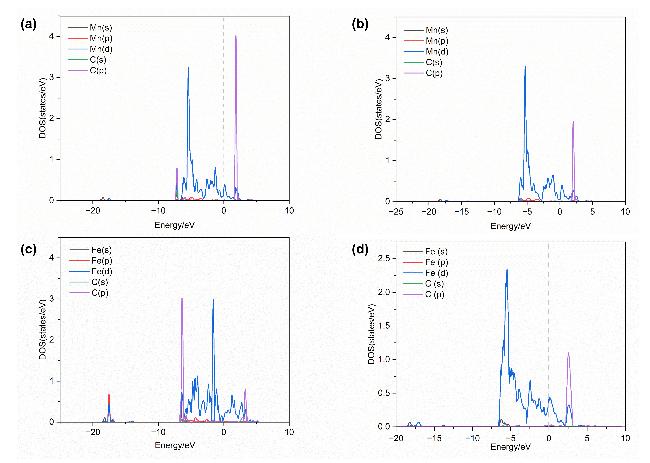
此外, 为了进一步了解NO在LaMnO3掺杂体系表面的吸附机理, 我们还分析了NO吸附在LaMnO3掺杂体系的PDOS. 如图9所示, N的2p轨道与Mn的d轨道之间出现了显著的杂化现象. 当N原子与Mn原子的d轨道杂化时, 吸附态会分裂为局部键合态和反键态. 具体而言, NO的5δ轨道向金属d轨道提供电子, 而金属d轨道则将电子反馈回NO的2π*轨道. 在这个过程中, Mn原子和N原子之间形成离子键或共价键. 图中的反键轨道均高于费米能级, 反键轨道不会被电子回流占据, 这有利于NO在催化剂表面吸附.
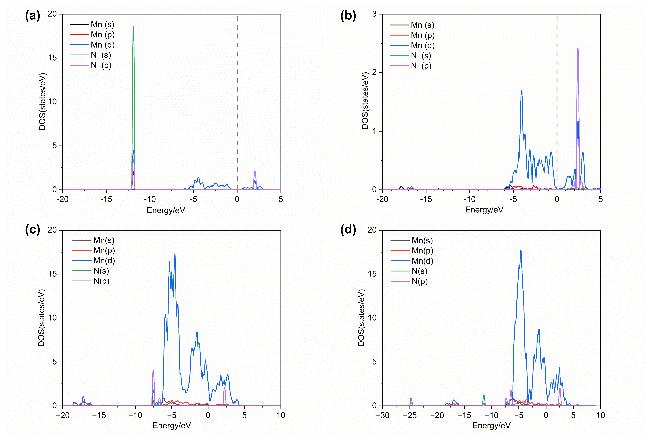
对于未掺杂构型, N的p轨道与Mn d轨道在–12.2 eV和2.4 eV处存在轨道杂化峰, 对于单掺杂Sr构型, N的p轨道与Mn的d轨道在2.5 eV处附近存在轨道杂化现象, 而–12.2 eV的轨道杂化现象消失. 实验结果表明Sr2+部分取代La3+后, Mn3+会变为Mn4+, 削弱NO的吸附强度, PDOS、吸附能及Bader计算结果与实验结果一致[29-30]. 相比之下, 单掺杂Fe构型和Sr-Fe共掺杂构型中, Mn d轨道与N p轨道在–10~–5 eV范围内发生强烈杂化, 在0~5 eV范围内, 两者存在共振峰, 表明NO分子与催化剂表面存在着强烈的相互作用. 值得注意的是, Fe掺杂显著提升了Mn d轨道峰值, 这主要归因于Fe掺杂有效调控了LaMnO3的电子结构, 增强了钙钛矿B位金属的电荷转移能力, 丰富了B位原子的价态, 并使Mn 3d轨道电子发生离域化, 加速NO的5δ轨道与金属d轨道的电子转移[21]. 此外, 与单掺杂Fe构型相比, Sr-Fe共掺杂构型中N的p轨道在–10~–5 eV区间的峰值明显降低, 进一步证实了Sr掺杂带来的晶格畸变和电荷补偿效应部分抵消了Fe掺杂所引起的电子增益效应. 晶格畸变可能导致局部电子结构的变化, 影响轨道重叠和电子转移效率, 从而调节NO的吸附行为.
2.5 CO氧化过程
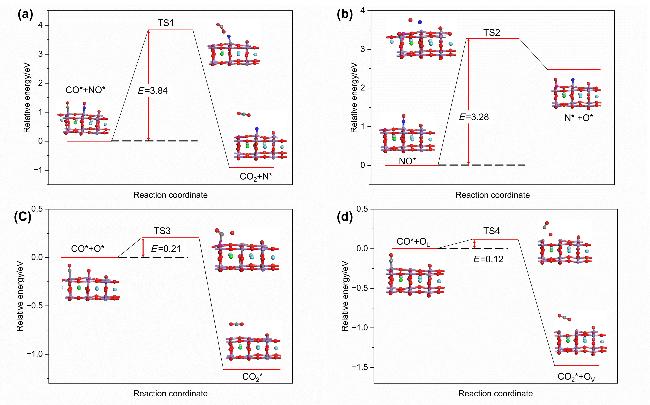
NO和CO同时吸附到催化剂表面, NO*的N—O断裂, 解离得到N*和O*, O*与吸附到Fe位点的CO结合生成CO2, 该反应克服了3.84 eV的活化能垒, 可知CO将NO直接解离成N*和O*, 生成CO2是非常困难的. 因此, CO直接还原NO生成N₂的路径在热力学与动力学上均不可行. CO最初吸附到表面的Fe位点, 与表面的晶格氧OL发生反应形成CO2, 克服0.12 eV的能垒, 形成过渡态, 然后CO2在催化剂表面脱附. 在SCR反应过程中, 活性O基团的存在会影响反应的结果, O活性基团主要来源于环境中O2分子的解离和N2O中间体的分解产生的O活性基团, 但两者不同来源的O活性基团对反应的影响也是不同的, 后续会对其进行具体分析.
在CO-SCR反应过程中, O2分子在催化剂表面经历解离, 其解离能垒为1.38 eV. 该过程导致O—O键断裂, 形成高活性的O自由基团. 这些O自由基随后分别吸附于催化剂表面的活性位点上. 与此同时, CO分子也吸附于相邻活性位点. 在后续的催化剂表面反应中, 吸附态的CO与解离产生的活性氧物种克服0.21 eV反应能垒发生氧化反应, 最终生成CO2. 上述反应一定程度上消耗CO-SCR反应过程中的还原剂, 从而降低了NOx的转换效率.
2.6 N2的生成过程
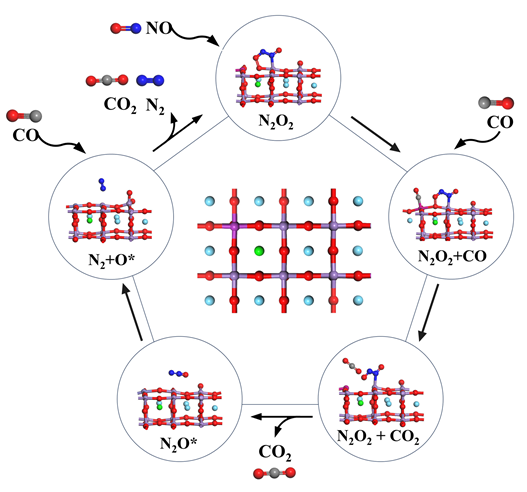
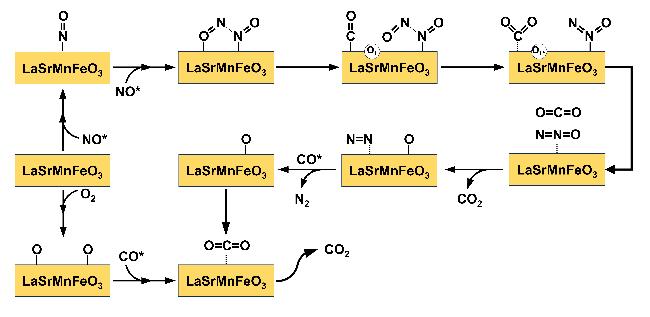
在研究CO在催化剂表面的主要氧化过程后, 进一步探讨了CO+NO在La1–xSrxMn1–yFeyO3催化剂表面的反应机理. 在CO选择性催化还原反应中, N2的生成可能有以下两种途径, (1) NO解离过程中吸附的两个N*原子的重新结合或CO直接还原NO; (2) N2O2中间体脱氧路径: 通过表面吸附的N2O2中间体解离形成N2.
为验证这两条路径的可行性, 因此, 首先计算了NO*→N*+O*和NO*+CO*→N*+CO2*的过渡态, 如图10a所示, 需要克服3 eV以上的能垒, 这表明NO直接解离和CO将NO直接解离成N*和O*是非常困难的, 并且两者很难促成N2的形成. 这可能源于两个NO分子吸附在表面中间体上形成N2O2*, 结合已有的研究表明: N2生成主要是通过N2O2*的脱氧反应形成的, 因此, 计算了N2O2*解离过程的反应能垒. NO倾向于N端与催化剂表面的Mn1原子结合, 吸附能为–1.79 eV, 属于强化学吸附. 另一个NO分子以O端向下吸附到Fe和Mn原子之间桥位的O原子/氧空位上, 形成N2O2*中间体. N2O2*中间体的N—N键长为0.116 nm. 该键长接近气相N2分子的0.109 nm, 显著短于自由N2O2*的0.1756 nm, 说明表面吸附显著促进了N—N键的形成. 此外, N和C同属于一个周期, 其原子序数大于C, 电负性强于C原子, 因此, N原子对N2O2*中间体左端O原子的电子引力强度大于C原子. 最后, N2O2*中间体通过破坏1个 Mn—N键和2个N—O键进行解离, 生成N2和吸附的O活性基团.
LaSrMnFeO3催化剂表面的反应能垒图如图11所示. 在上述反应过程中, 气相CO分子首先吸附在催化剂表面Fe位上, 该吸附过程为放热反应, 吸附能为 –0.63 eV, 属于弱化学吸附. CO捕获催化剂表面的晶格氧OL发生氧化反应, 生成CO2, 其反应能垒为0.12 eV, 由于CO2与催化剂表面结合较弱, CO2快速脱附并生成一个氧空位OV. N2O2*中间体在桥位的N—O键断裂后, 解离产生的OL填补氧空位OV. 桥位吸附的N—O键断裂后, 新生成的OL*填补氧空位, N端与Mn位NO的N原子结合形成N2O*中间体, 反应能垒为0.69 eV. 随后, 气相的中间体N2O*吸附在Mn/Fe催化剂表面并克服1.14 eV反应能垒, 破坏N—O键生成N2和O*. 同时, 产物N2的吸附能较低, 易于释放. N2O*中间体解离生成的活性O基团倾向于吸附到催化剂表面, 增加与气相CO分子接触的机会, 从而容易发生氧化反应. 该反应过程克服了0.21 eV的活化能垒, 释放了大量的反应热(1.16 eV), 在反应动力学和热力学上具有巨大优势. 这有利于推动N2O*中间体分解反应平衡向右移动, 加快N2O*中间体的分解速度, 最终转变为N2, 减少N2O*副产物的产生. 这与现有的理论和实验研究结果相吻合[33-35].

在上述反应过程中, N2O*→TS3→N2+O*被确定为上述反应途径的限速步骤, 因为该步骤具有最高的活化能垒. 对比未掺杂LaMnO3催化剂的复杂双分子反应机制, Sr-Fe共掺杂促使LaMnO3催化剂表面CO-SCR核心反应机制向Mars-van Krevelen机制转变, 与实验结果相符合, 其中关键速控步骤N2O*→N2+O*的反应能垒较未掺杂的双分子反应机制[7]的1.64 eV显著降低至1.14 eV, 有利于反应的进一步进行.
2.7 Sr/Fe掺杂对LaMnO3抗硫、抗水中毒性能的影响
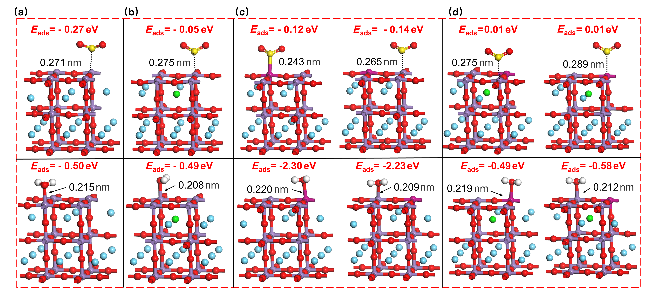
在实际应用的选择性催化还原反应过程中, H2O和SO2等气体组分可能引发催化剂表面钝化及活性位点覆盖, 导致催化剂中毒失活. 因此, 进一步探究催化剂的抗毒机理也至关重要, 尤其是抗水中毒和抗硫中毒方面, 本文优化了掺杂体系催化剂表面SO2和H2O的吸附构型, 计算了其在LaMnO3掺杂体系表面主要核心活性位点的吸附能, 吸附构型及吸附能如图12所示.
2.7.1 抗硫性能
计算结果表明: 在未掺杂及Sr/Fe单掺杂、共掺杂构型中, SO2在催化剂表面主要的活性位点的吸附能显著低于CO和NO在催化剂表面的吸附能, 表明SO2与CO/NO的吸附存在热力学竞争差异. 在催化反应过程中, CO/NO因其更强的吸附能倾向优先占据活性位点, 而SO2难以有效吸附, 从而显著降低了硫中毒风险. 值得注意的是, 单掺杂Sr或Fe原子后, SO2在催化剂表面的吸附能较未掺杂LaMnO3显著降低, 表明Sr和Fe原子的引入有效改善了LaMnO3催化剂的抗硫中毒的性能, 这与实验现象表现一致[38-39]; 此外, Sr-Fe共掺杂构型的SO2吸附能Eads=+0.01 eV进一步趋近于非自发区间, 表明Sr/Fe掺杂的协同调控, 抑制了气相SO2在催化剂表面的吸附, 表现出优异的抗硫中毒性能.
2.7.2 抗水性能
在CO-SCR反应过程中, H2O分子通常与CO和NO发生竞争性吸附, 占据催化剂表面活性位点, 导致催化剂失活. H2O在LaMnO3催化剂表面Mn位点的吸附能为–0.50 eV, 在LaSrMnO3催化剂表面Mn位点的吸附能为–0.48 eV, 在LaMnFeO3催化剂表面Mn和Fe位点的吸附能分别为–2.23和–2.30 eV, 在LaSrMnFeO3催化剂表面Mn和Fe位点的吸附能分别为–0.58和–0.49 eV. 由此可见, 单掺杂Sr原子对H2O分子在LaMnO3催化剂表面的吸附起到了轻微抑制作用, 单掺杂Fe原子加剧了H2O分子在催化剂表面的吸附, 形成稳定的化学吸附构型, 导致催化剂水中毒失活. 这是因为Fe的电负性强于Mn, 因此Fe3+的Lewis酸性强于Mn3+, Fe会优先吸附H2O分子并促进它解离(H2O→H++OH–). 邻近的Mn位点由于Fe的掺杂发生氧化还原反应(Mn3++Fe3+→Fe2++Mn4+), 促进了电子向未占据的d轨道转移, 有利于在表面形成稳定的OH–, 促使H2O分子在催化剂表面的解离性吸附. 而引入Sr的掺杂有助于进一步抑制H2O的竞争性吸附, H2O吸附从在单掺杂Fe催化剂表面的强化学吸附进一步转换成物理吸附. 这是由于Sr的掺杂使Fe3+难以还原为Fe2+, 抑制电子转移, 降低了其氧化还原能力, 同时Sr掺杂也使晶格刚性增加, 限制了表面原子弛豫, 不利于H2O分子后续的解离性吸附.
综上所述, 单掺杂Sr原子可以同时对LaMnO3催化剂抗硫中毒/抗水中毒性能进行轻微改善, 单掺杂Fe原子能够有效抑制SO2在催化剂表面的化学吸附, 但会加剧H2O分子的不可逆吸附导致催化剂中毒失活. Sr-Fe共掺杂催化剂对SO2和H2O的吸附能显著低于对CO/NO的吸附能, 表明Sr-Fe共掺杂的协同调控作用能够提高LaMnO3催化剂的抗水中毒和抗硫中毒性能.
2.8 主要反应路径
在LaSrMnFeO3催化剂表面模拟了CO氧化和 CO+NO反应过程, 如图13所示. 反应机理如下:
2CO+2NO→N2+2CO2
CO+OL→CO2+OV
O2→2O*
CO+O*→CO2
NO+OV→N2O2*→N2O*+OL*
N2O*→N2(ad)+O*
式中OL表示催化剂表面的点阵氧, OL*表示催化剂表面的补充形成的晶格氧, OV表示CO氧化反应后表面形成的氧空位.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
本文基于密度泛函理论(DFT)从几何结构、吸附能、电子性质等方面系统研究了LaMnO3掺杂体系的CO选择性催化还原反应机理, 进一步探讨了Sr/Fe掺杂对LaMnO3催化活性及抗硫、抗水中毒性能的影响.
(1)在LaMnO3掺杂体系中, 发现Sr和Fe的掺杂增强了催化剂对NO和CO的吸附能力. 结果表明, Sr和Fe的掺杂引起了电子迁移和表面晶格位错, 这种现象增强了表面酸性位点的活性, 促进了NO和CO分子在催化剂表面的吸附. 此外, 筛选出两个较优的吸附位点: NO倾向于在N端吸附于Mn位点, 而CO在Fe掺杂体系中更易通过C端吸附于Fe位点.
(2)阐明了Sr/Fe掺杂对LaMnO3催化剂抗硫和抗水中毒性能的影响. 计算结果表明, 单掺杂Sr原子可以轻微改善LaMnO3催化剂的抗硫和抗水中毒性能, 而单掺杂Fe原子则能够有效抑制SO2在催化剂表面的化学吸附, 但会加剧H2O分子的不可逆吸附, 导致催化剂中毒失活. Sr-Fe共掺杂催化剂对SO2和H2O的吸附能显著低于对CO和NO的吸附能, 表明Sr-Fe共掺杂的协同调控作用能够提高LaMnO3催化剂的抗水中毒和抗硫中毒性能.
(3)揭示了LaSrMnFeO3催化剂表面的CO-SCR反应机理. CO与催化剂表面的晶格氧发生反应, 生成CO2并形成氧空位. NO形成的关键中间体N2O2中的N—O键断裂以填补氧空位, 符合Mars-van Krevelen反应机理. N端的NO与Mn位结合形成N2O中间体, 随后N—O键的破坏生成N2和O*. 最终, 通过CO的氧化反应消耗O*, 加速N2O的分解反应, 减少N2O副产物的积累. N2O*→N2+O*是NO在LaSrMnFeO3表面CO-SCR反应的限速步骤, 其活化能垒从未掺杂催化剂的1.64 eV降低至1.14 eV, 有利于反应的加速进行. 此外, 在富氧环境下, O2的解离产生活性氧基团会优先吸附在催化剂的表面活性位点, 与CO反应消耗还原剂, 从而抑制CO-SCR反应的进行.
4 计算方法
催化剂模型通过Materials Studio 2018软件构建, 采用基于密度泛函理论的VASP软件包进行第一性原理计算, 并结合VASPKIT程序包实现元素赝势组合与数据后处理. 采用了PAW-PBE交换泛函描述交换关联势. 通过收敛性测试, 选用ENCUT取值为520 eV的平面波扩展. 在布里渊区的积分过程中, 采用Gamma方案生成K空间网格, 选取K点网格大小为3×3×3. 采用Hubbard的DFT+U方法精确地修正了Mn 3d和Fe 3d态的强原位库仑相互作用. 对于Mn 3d, U值选择为3.9 eV, 对于Fe 3d, U值选择为3.4 eV. 本文计算的收敛容限定义如下: 最大受力为0.3 eV/nm, 总能量为1.0×10–5 eV/atom, 位移为0.0001 nm, 自洽场为1.0×10–6 eV/atom. 优化后的LaMnO3钙钛矿单胞模型晶胞, 其晶格常数变为a=b=c=0.390 nm, α=β=γ=90°, 与本课题组实验值a=b=c=0.388 nm, α=β=γ=90°基本一致. 因此, 本文使用的计算方法是可靠的.
吸附能计算的表达式为:
Eads=Etotal–Eslab–Eadsorbent
式中Eads为吸附能(eV); Etotal为吸附体系总能量(eV); Eslab为基底能量(eV); Eadsorbent为吸附小分子能量(eV). 吸附能的计算值能够揭示吸附过程的进行难易及其热力学稳定性特征. 一般, 若吸附能呈现负值, 表明吸附过程是一个放热反应, 吸附稳定. 相对地, 当吸附能为正值时, 则意味着吸附过程是吸热的, 吸附不稳定.
反应途径由初始态(IM)、过渡态(TS)和终态(FS)三部分构成. 采用VASP中的CI-NEB方法搜索并确定了催化剂表面CO-SCR反应途径, 该方法被认为是一种计算过渡态结构的有效方法. 每个过渡态的计算都显示出显著的虚频率值(f/i), 证实了结果的可靠性. 活化能反应势垒(Ea)的定义如下:
Ea=ETS–EIM
其中Ea表示反应能垒, ETS表示过渡态的能量, EIM表示反应物的能量.
(Cheng, F.)