

1 引言
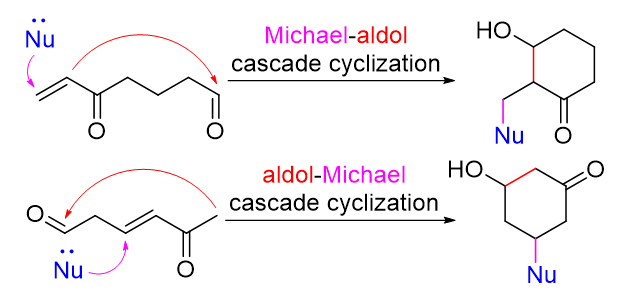
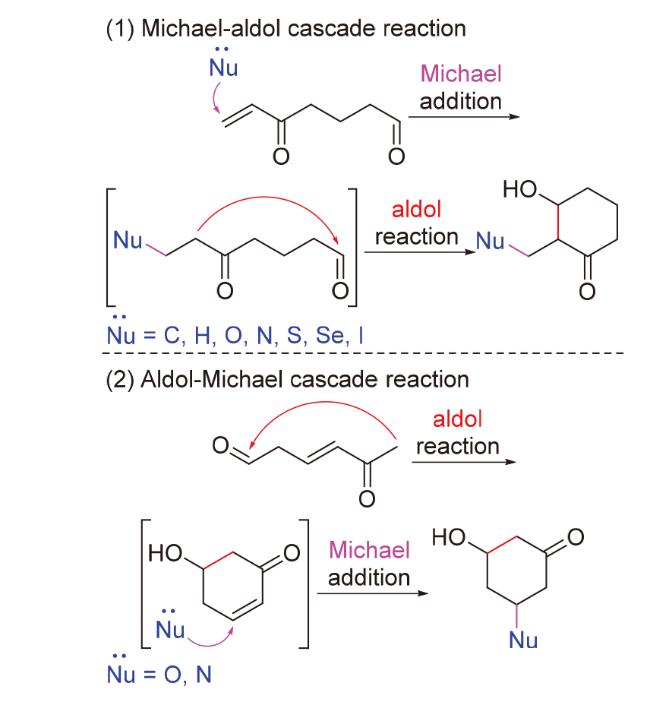
Michael加成[3-5]是多种亲核试剂对π电子体系(共轭烯酮等)的1,4-加成反应. Aldol反应[6-9]是烯醇(盐)对醛和酮的加成反应. 二者均围绕“羰基”这一重要官能团发生, 故常适合被设计为串联反应, 以提高合成效率, 简化步骤. 例如: Michael加成中间体可进一步与分子内的醛、酮位点发生aldol反应, 构建复杂环系(Michael- aldol串联反应, Scheme 1); 亦或是, aldol产物中的共轭烯酮可通过进一步的Michael加成引入新的所需原子, 构建新的手性中心(aldol-Michael串联反应, Scheme 1). 其中, 最著名的经典应用案例莫过于利用环酮与共轭烯酮反应得到环烯酮的Robinson环化反应[10-11]. 这些反应在针对结构复杂、来源稀缺及具有重要生理活性的天然产物的全合成工作中应用广泛, 但尚未有过系统性的综述报道. 基于上述两种串联反应, 本文在Michael加成方面, 按照不同的亲核试剂类型进行分类; 在aldol反应方面, 侧重关注分子内aldol反应, 以介绍Michael/aldol串联环化反应作为关键步骤在天然产物合成中的应用.
2 Michael-aldol串联反应
2.1 C-Michael-aldol串联反应
C-Michael-aldol串联反应多用于构建复杂的环系结构. 可通过有机小分子胺催化醛酮生成烯胺或碱攫取醛酮α-H生成烯醇盐的途径原位制备高活性C-亲核试剂, 对共轭体系或其它醛酮位点进行加成. 其中, 有机小分子胺多见脯氨酸及其衍生物和金鸡纳碱及其衍生物, 包括奎尼丁(quinidine)衍生物和奎宁(quinine)衍生物. 它们在控制反应的立体选择性方面发挥重要作用.
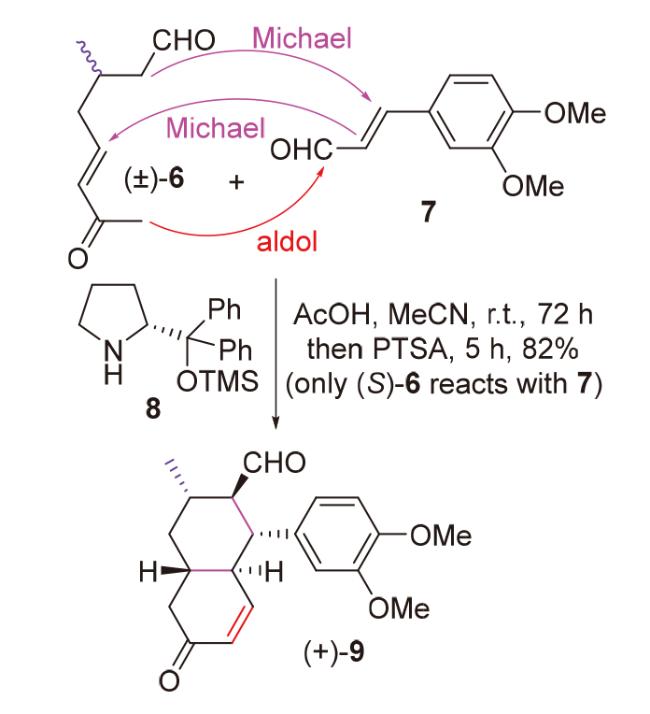
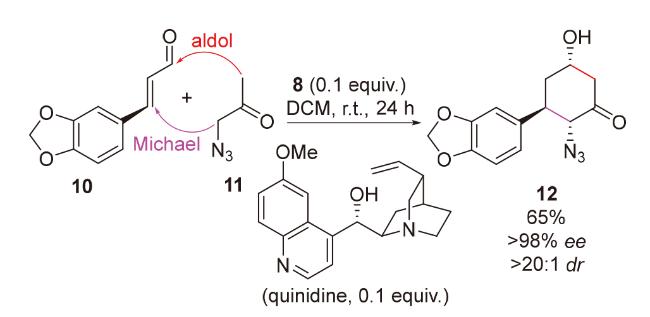
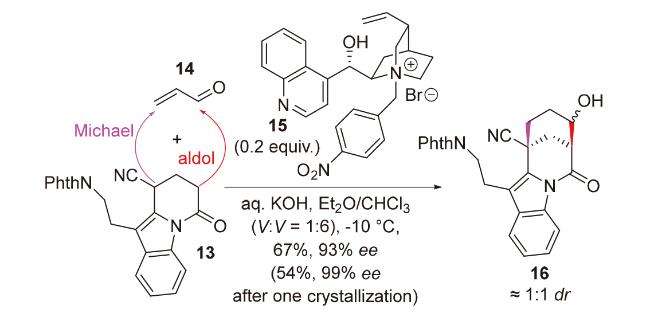
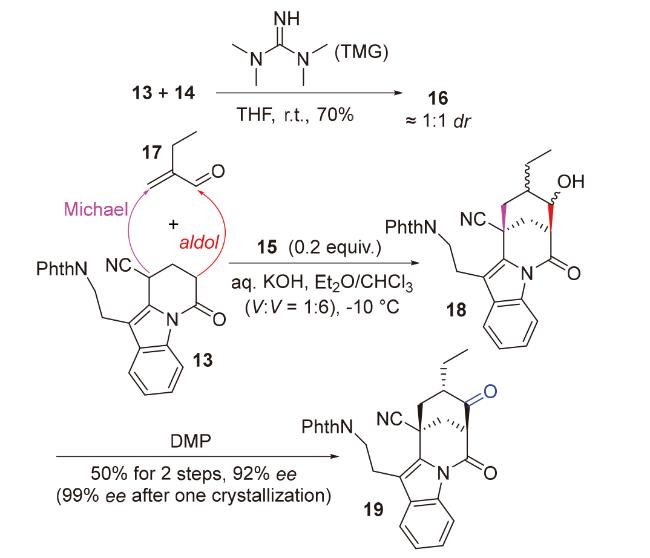
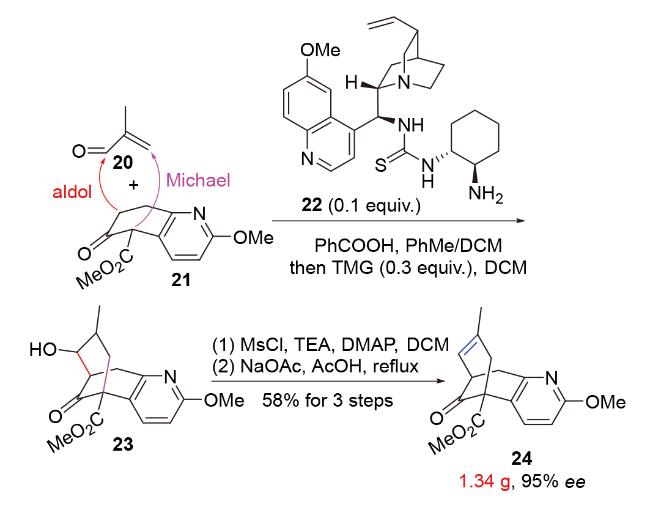
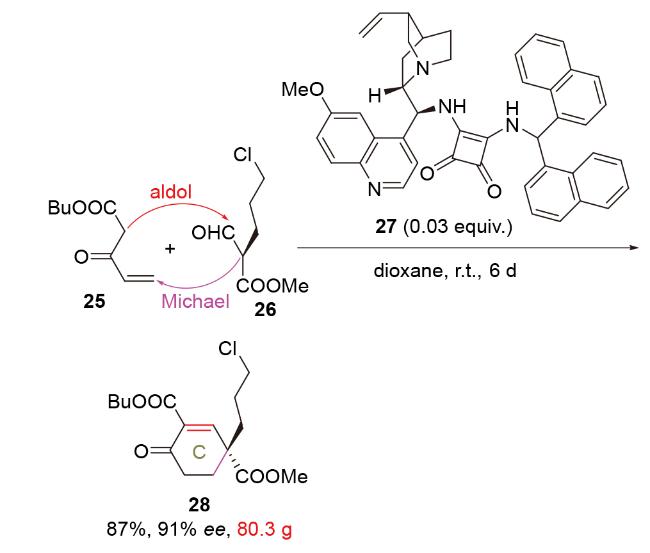
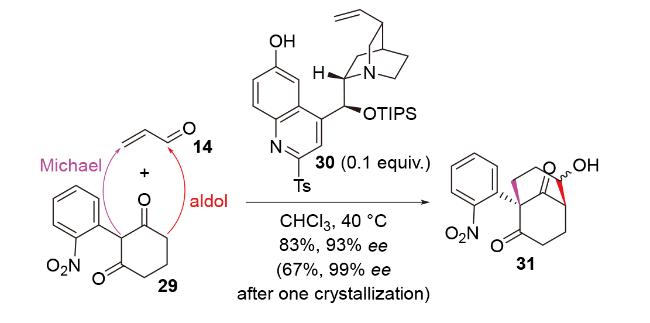
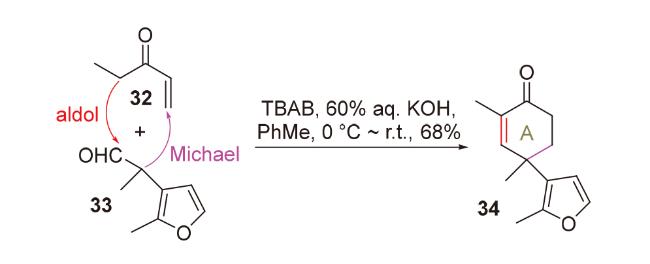
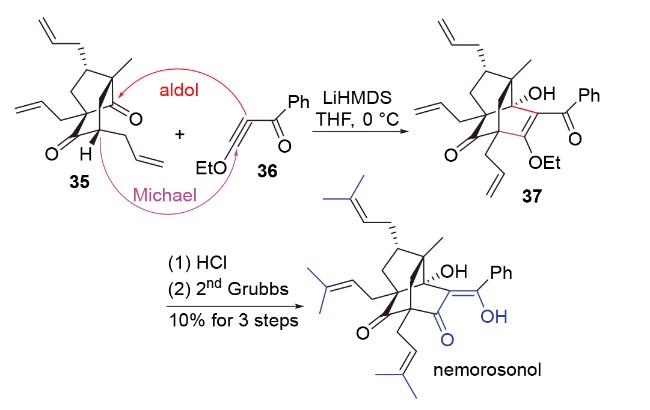
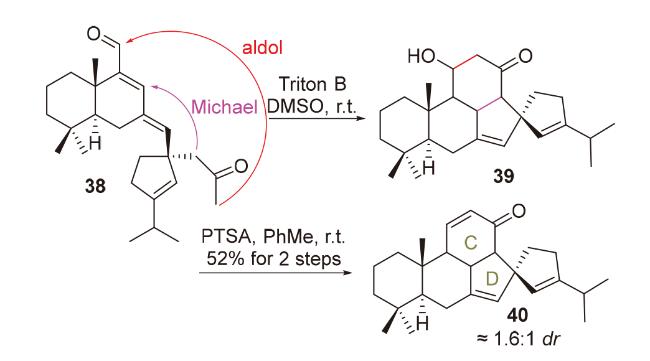
2020年, 韩福社团队[17]在对chippiine-dippinine家族的其它4个成员(+)-demethoxychippiine、(-)-dippinine B、(+)-dippinine C和(-)-demethoxychippiine的全合成中, 发现碱四甲基胍(TMG)亦可促进上述相同反应的发生, 再次合成了桥环结构16; 在对该家族的其它3个成员(-)-demethoxychippiine、(-)-3-O-methyldemethoxy- chippiine和(-)-3-hydroxy-3,4-secocoronaridine的全合成中, 再次利用了催化剂15以构建桥环结构18 (Scheme 7).

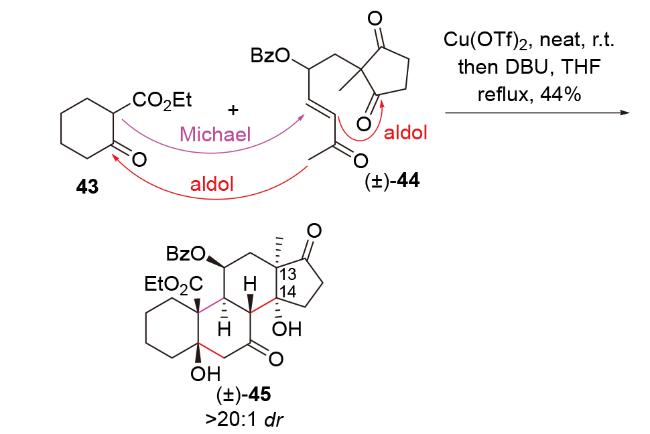
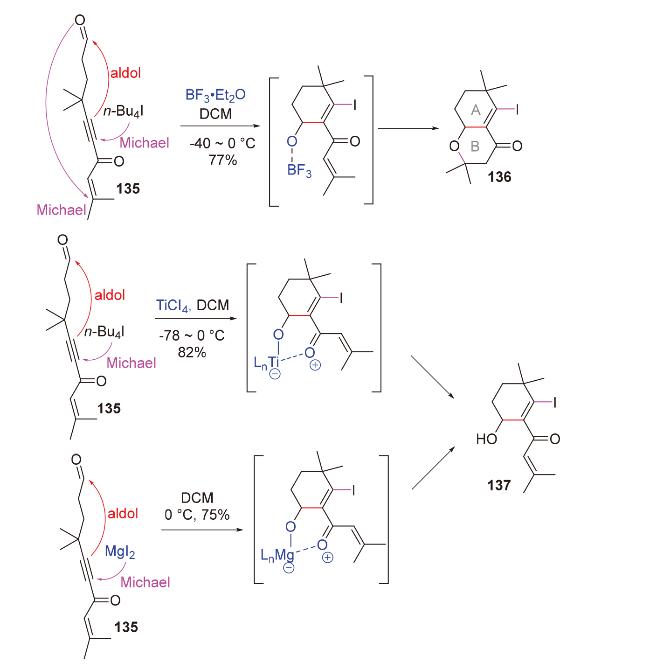
路易斯酸亦可促进C-Michael-aldol串联反应. 2016年, Nagorny团队[26]在甾体类心肌细胞钠/钾ATP酶抑制剂cardiotonic家族中的19-hydroxysarmentogenin和trewianin aglycone的全合成中, 以C-Michael-aldol-aldol串联反应为模型反应, 在未添加手性配体的情况下, 高立体选择性地构建了该类分子的核心骨架(±)-45 (Scheme 15). 其中, Cu(OTf)2诱导了Michael加成, 原料(±)-44中的γ-OBz手性中心高效控制了Michael加成的立体选择性. 不足的是, 在1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)促进的两步aldol环化反应中, 其中一步构建的C(13)和C(14)位立体中心与天然产物所需相反, 迫使作者后续需要通过双三甲基硅基胺基钠(NaHMDS)促进的retro aldol-aldol反应进行调整.
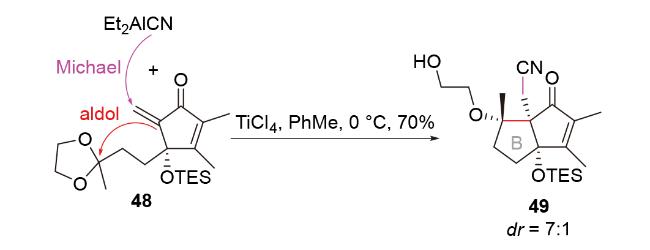
2.2 H-Michael-aldol串联反应(还原aldol反应)
虽然, 负氢试剂作为亲核试剂对共轭烯酮的1,4-加成反应通常被称为“1,4-还原”或“共轭还原”, 而非传统意义上的Michael加成; 但其亦可原位生成烯醇负离子, 进而与醛酮发生aldol反应——这一串联反应又被成为“还原aldol反应”. 这类反应具有传统aldol反应不具有的一些优点: (1)体系温和, 酸碱性弱, 可避免包括retro aldol、β-消除在内的一系列副反应; (2)反应位点可控, 可区域选择性地生成烯醇负离子. 因而在天然产物合成中应用广泛.
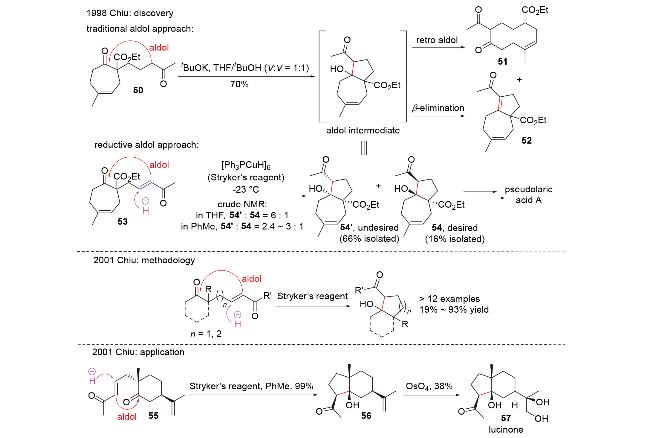
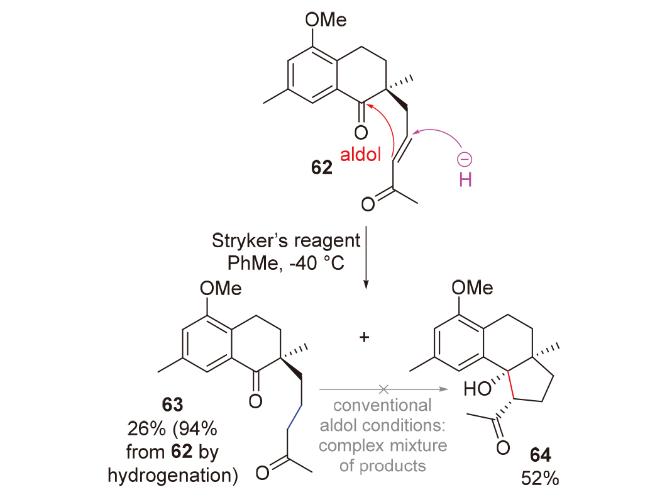
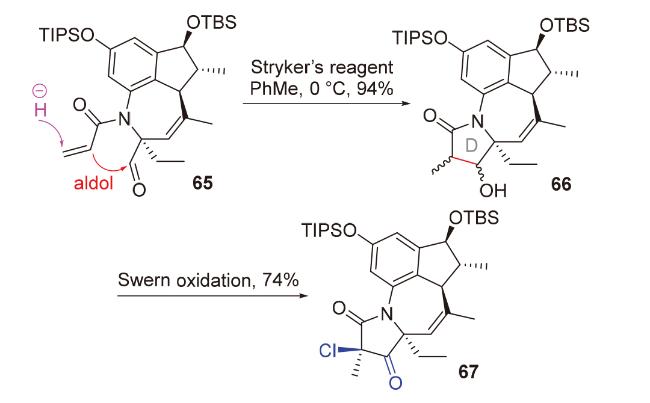
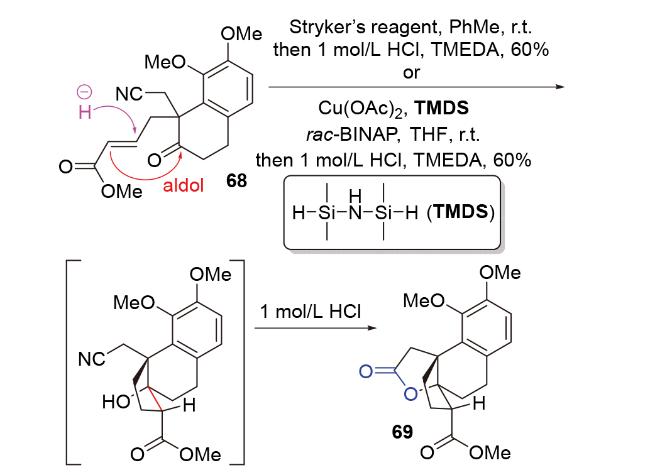
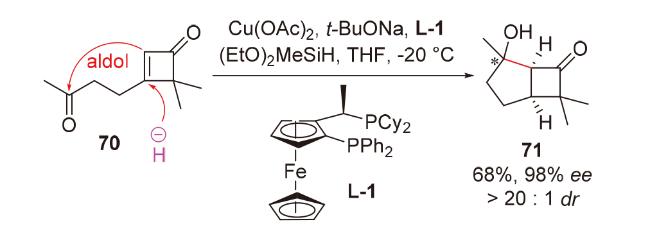
1998年, Chiu团队[29]在具有抗癌、抗真菌活性和小鼠避孕效用的二萜pseudolaric acid A的合成研究(Scheme 18)中, 尝试使用传统的碱促进的分子内aldol反应构筑目标分子的并环骨架. 然而, 由于体系的碱性, aldol产物会直接生成retro aldol产物51和β-消除产物52, 导致其无法被成功制备. 为了避免碱性体系, 他们开创性地提出了负氢试剂对共轭烯酮1,4-还原以原位产生烯醇负离子, 进而发生分子内aldol反应的思路. 他们使用非常温和的、具有极佳区域选择性和化学选择性的铜氢类试剂——Stryker试剂, 在低温下处理烯酮53, 顺利得到具有pseudolaric acid A核心骨架的aldol产物54'和54, 且完全未观察到副产物51和52; 将溶剂从四氢呋喃改为非配位溶剂甲苯, 可以提高所需构型产物54的比例. 基于此, 2001年, 他们对这类还原aldol反应进行了系统性的方法学研究(Scheme 18)[30], 并成功利用该反应为关键步骤完成了倍半萜lucinone (57)的全合成(Scheme 18)[31].
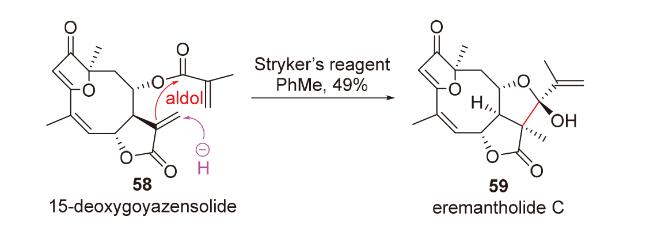
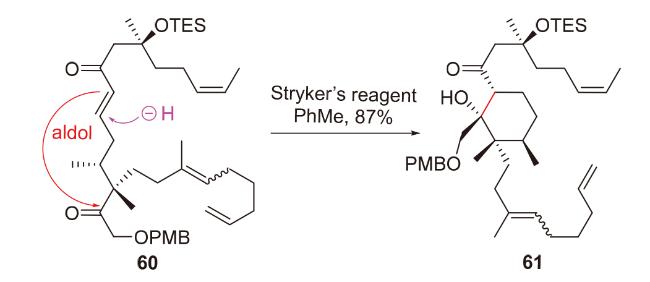
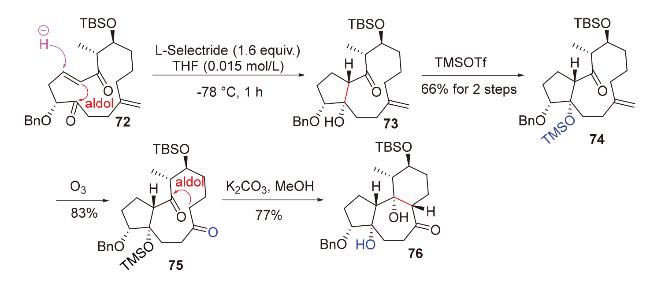
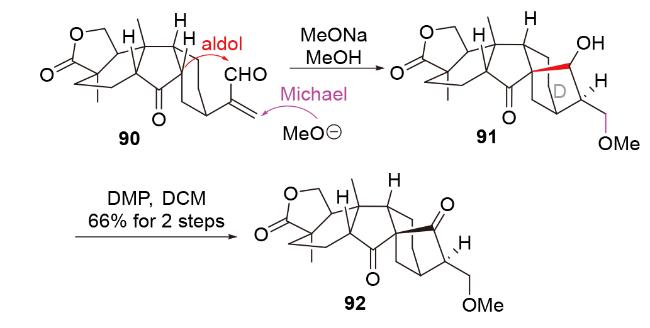
2.3 O-Michael-aldol串联反应
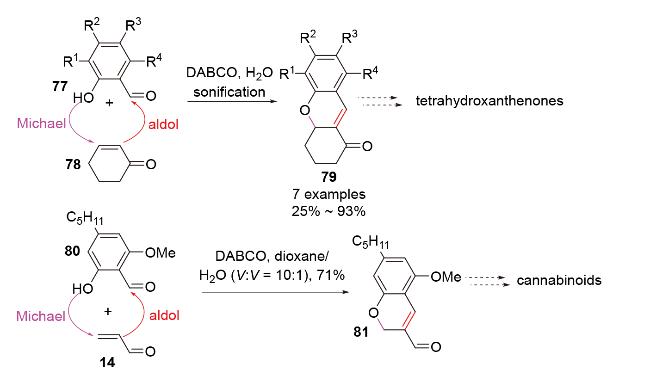
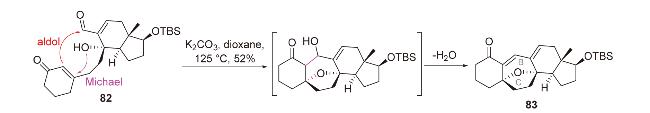
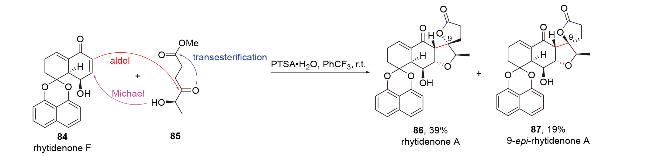

2.4 N-Michael-aldol串联反应
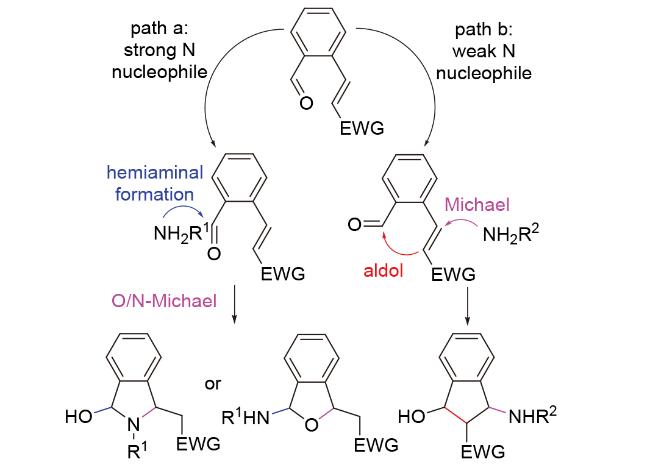
虽然N-Michael加成反应在合成中应用广泛, 但相比于其他种类原子的Michael-aldol串联反应, N- Michael-aldol串联反应却鲜有报道, 这是因为N原子本身性质特殊. 2013年, 孙建伟团队[47]在发展用于合成aminoindanol类化合物的N-Michael-aldol串联反应方法学时指出: 对于大多数胺, 由于其较强的亲核性和醛酮较强的亲电性, 将存在优先进攻醛酮而非烯烃, 形成半胺醛, 并进一步发生分子内O/N-Michael加成的竞争反应(Scheme 31, path a); 因此, 若想确保N-Michael-aldol串联反应顺利发生, 应选用亲核性较弱的N类亲核试剂, 如磺酰胺等(Scheme 31, path b).
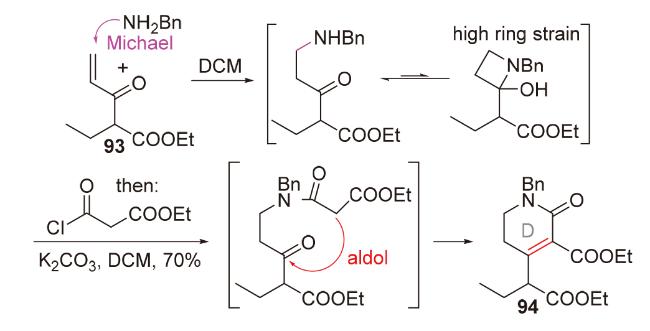
2.5 S-Michael-aldol串联反应
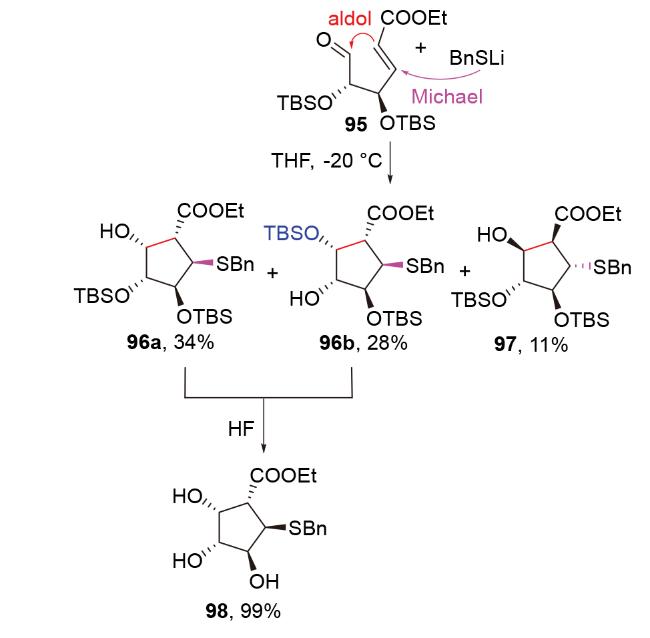
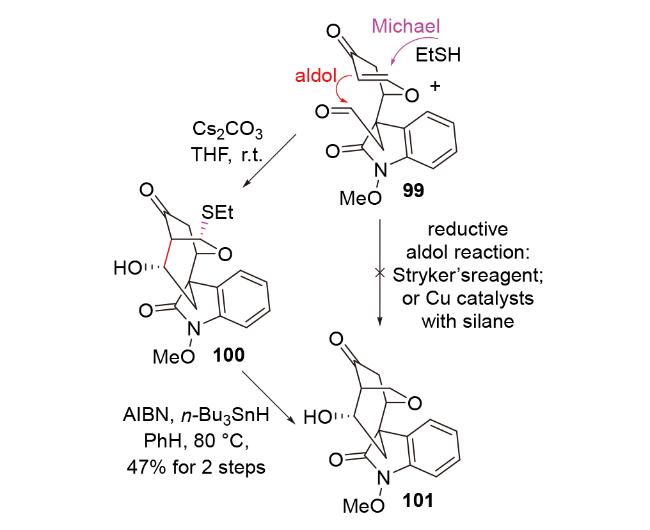
2016年, 赵刚团队[50]在钩吻类单萜吲哚生物碱gelsedilam的全合成中, 试图利用负氢试剂参与的还原aldol反应构建目标分子的高张力笼状[3.2.2]桥环骨架101, 并未成功. 他们认为, 原料99结构特殊——其烯烃部分为烯酮与烯醇醚共享, 导致了该烯烃较弱的亲电性, 因此很难被亲核性本就较差的负氢试剂进攻, 以引发还原aldol反应. 于是, 他们改用亲核性更强的乙硫 醇/碳酸铯组合, 成功引发了所需的S-Michael-aldol串联反应. 其产物100经还原脱硫, 进一步转化为所需的骨架结构101 (Scheme 34). 该案例说明, S-Michael-aldol串联反应与脱硫反应的组合可被视为还原aldol反应的替代反应, 具有借鉴意义.
2.6 Se-Michael-aldol串联反应
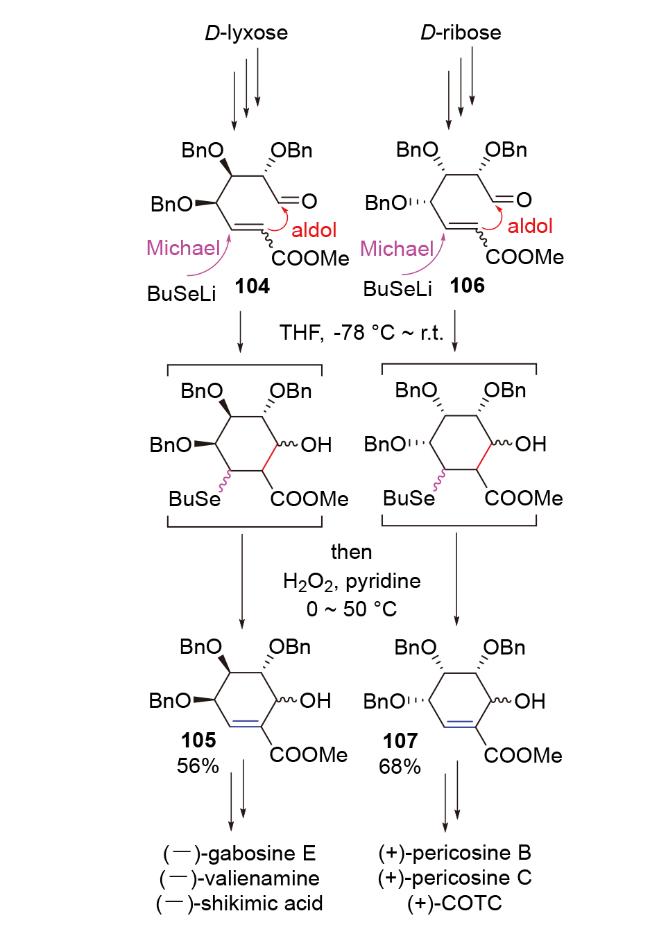
2019年, Buda团队[52]在具有多种重要生理活性的carbasugar类天然产物(-)-gabosine E, (-)-valiena- mine和(-)-shikimic acid的合成研究中, 从D-来苏糖出发, 利用硒基锂试剂引发的Se-Michael-aldol-氧化消除串联反应, 合成了该类分子的环己烯骨架105 (Scheme 36). 2020年, 他们[53]又利用该反应, 从D-核糖出发, 合成了carbasugar类天然产物(+)-pericosine B, (+)- pericosine C和(+)-COTC的环己烯骨架107, 进而完成了它们的全合成(Scheme 36).
2.7 I-Michael-aldol串联反应
2018年, 高栓虎团队[54]发表了综述《Halo-Michael/ Aldol串联反应及其在合成中的应用》. 该综述中, 虽然Cl原子和Br原子参与的此类反应有被提及, 但在涉及天然产物合成的应用部分, 仅有五个I-Michael-aldol串联反应的应用案例被介绍.
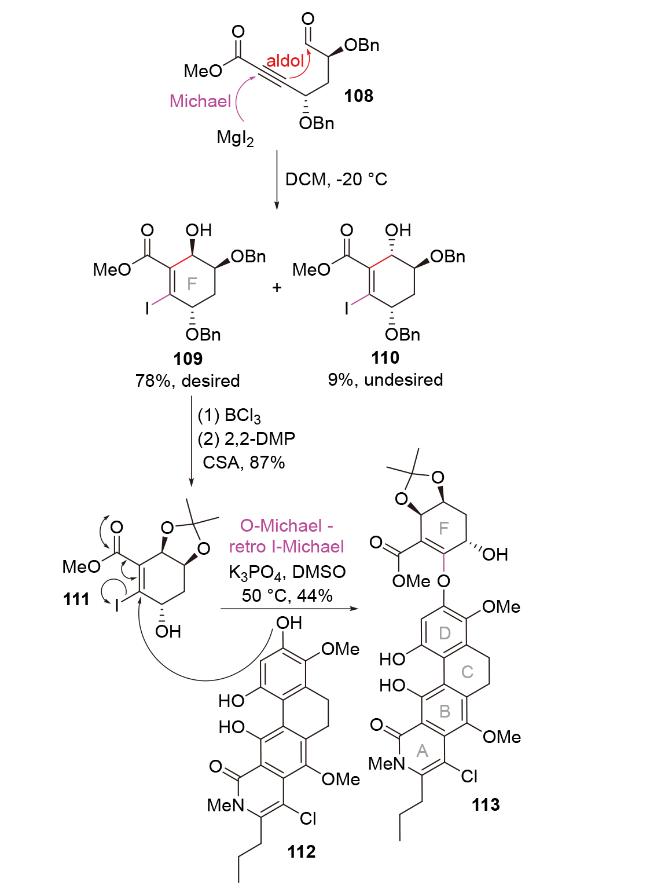
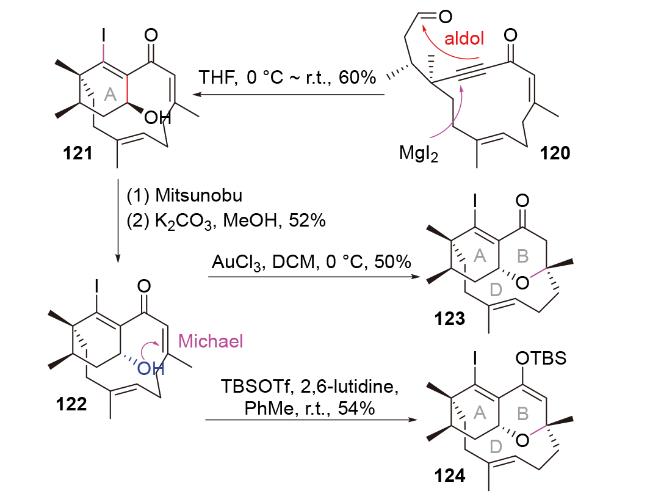
2011年, Porco团队[55]在具有广谱抗癌活性的聚酮天然产物kibdelone C的全合成中, 利用碘化镁促进的I-Michael-aldol串联反应, 合成了该分子的碘代F环结构109 (Scheme 37). 反应的立体选择性由镁离子与醛基氧原子、α-苄氧基氧原子之间的螯合作用控制. 后续, 他们巧妙地利用衍生产物111和酚112之间的O- Michael-retro I-Michael反应, 合成了五环结构113. 这不仅实现了目标分子F环和A/B/C/D环之间的组装, 同时完成了对碘原子的拆卸, 也保留了目标分子F环中所需的烯烃官能团. E环可进一步通过分子内Friedel- Crafts酰化反应形成.
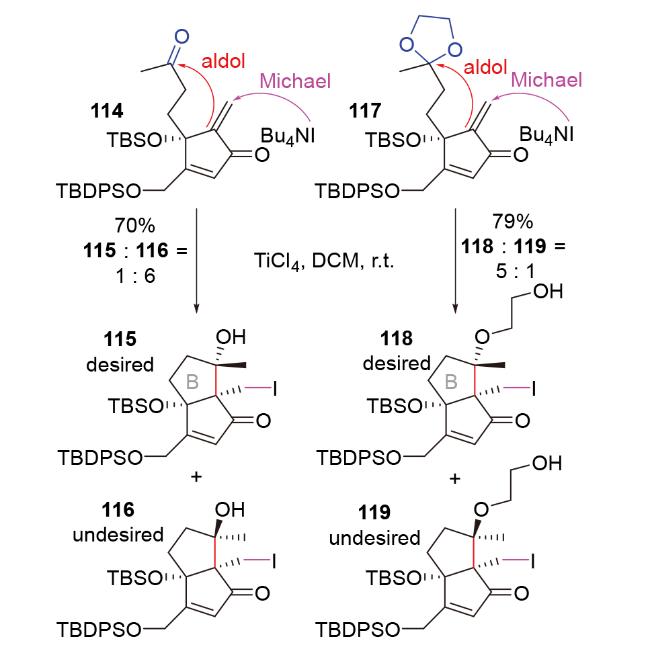
2012年, Greaney团队[28]在倍半萜merrilactone A的形式合成中, 利用四丁基碘化铵为碘源的I-Michael- aldol串联反应, 构建了该分子的B环结构115 (Scheme 38). 然而, 一方面, 所需构型产物115所占比例较小; 另一方面, 两种产物115和116在后续诸多反应条件下不稳定, 极易发生retro aldol副反应. 为避免此副反应, 他们设计了以缩酮为aldol亲电体的结构117以进行Michael-aldol串联反应, 也成功实现了所需转化, 并大幅提升了所需构型产物118所占比例. 遗憾的是, 由于围绕产物118中碘官能团的后续转化均宣告失败, 他们最终改用氰基铝试剂参与的Michael-aldol串联反应(Scheme 17), 完成了目标分子的形式合成.
3 Aldol-Michael串联反应
3.1 Aldol-O-Michael串联反应
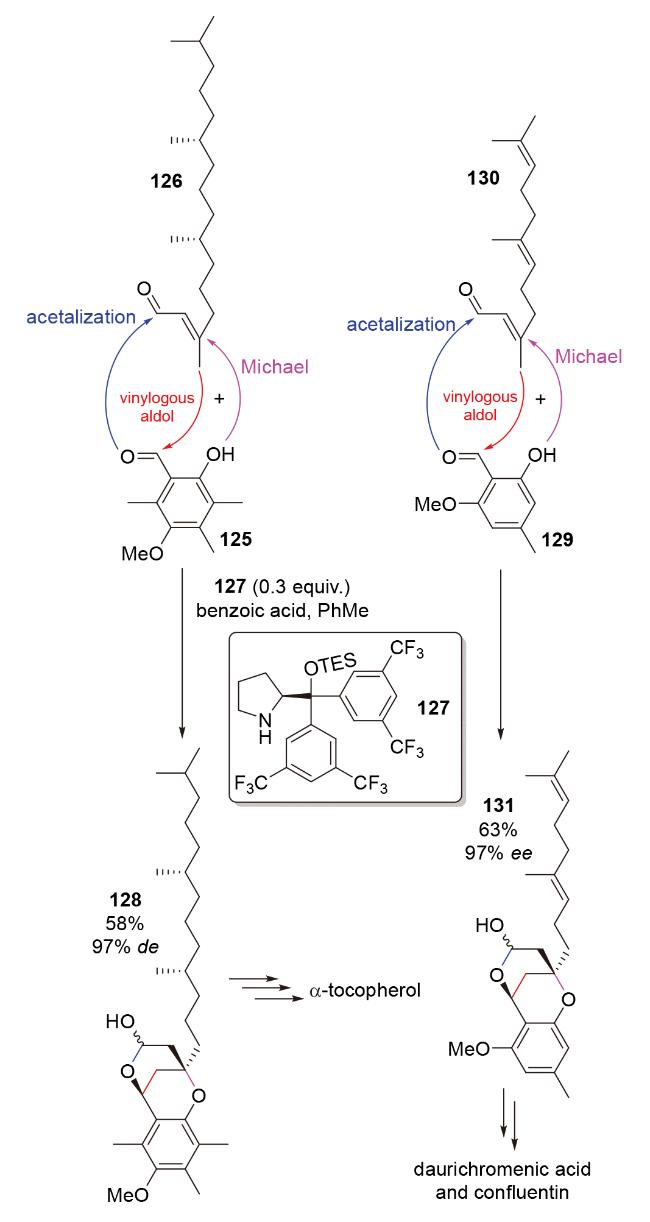
2008年, Woggon团队[57]在具有自由基断链抗氧化功能的vitamin E家族chromanol类天然产物α-toco- pherol的全合成中, 利用脯氨酸衍生物127催化的插烯aldol-O-Michael-缩醛化串联反应, 构建了目标分子的核心骨架结构128 (Scheme 40). 在机理方面, 该反应经历了烯醛126衍生烯胺中间体对醛125的插烯aldol反应; 随之生成的共轭烯亚胺与酚发生分子内Michael加成; 最后, aldol产物醇与亚胺水解得到的醛生成半缩醛128. 2010年, 该团队[58]再次利用该反应, 完成了具有强效抗HIV活性的chromene类天然产物daurichromenic acid和具有人香草酸受体VR1拮抗活性的chromene类天然产物confluentin的全合成(Scheme 40).

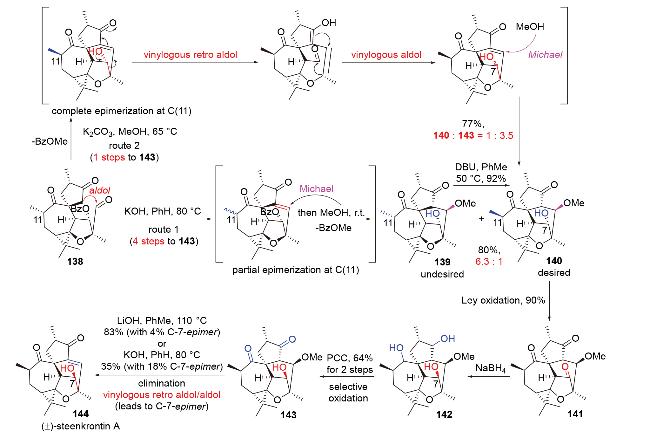
2015年, 丁寒锋团队[60]在二萜steenkrotin A的全合成中, 利用三酮138的分子内aldol缩合反应, 构建了全合成中目标分子的最后一个六元环, 分为两条路线(Scheme 43). 由于反应体系为碱性的甲醇溶液, 分子内aldol缩合反应还伴随着C(11)位甲基部分差向异构化-去苯甲酰化-O-Michael加成的串联反应. 与高栓虎的工作(Scheme 29)类似, 产物139和140中的甲氧基是多余的, 需要后续通过E1CB消除反应来剔除. 在路线1中, 所需的产物140占比较低, 需要进一步通过用碱DBU异构化主产物139来大量制备. 随后, 他们通过氧化-还原策略, 翻转了C(7)位的羟基立体中心, 以线性4步, 合成了化合物143. 然而, 在最后一步碱性下的E1CB消除反应中, 他们意外发现, 刚刚被修正的C(7)位立体中心会再通过插烯型retro aldol-aldol串联反应发生消旋, 导致生成一部分C(7)-epi-steenkrotin A. 受此现象启发, 在路线2中, 他们巧妙设计出了三酮138的分子内aldol缩合-C(11)位甲基完全差向异构化-去苯甲酰化-插烯型retro aldol-aldol-O-Michael串联反应, 将化合物143的合成路线从之前的4步缩短到了1步.
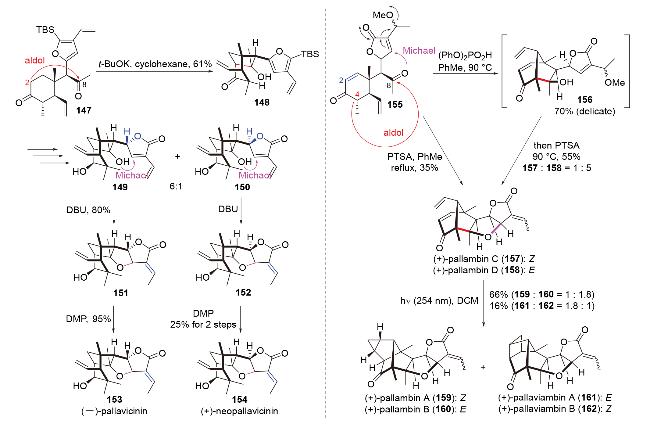
2024年, 贾彦兴团队[62]利用基于仿生aldol反应的骨架多样化策略, 完成了八个苍白球菌(pallavicinia)二萜的多样性全合成(Scheme 45). 他们先是在碱性条件下实现二酮147的C(2)-C(8)跨环aldol反应, 构建了桥环骨架148; 148经一系列转化得到非对映异构体烯酯149和150; 149和150在碱性下发生O-Michael-双键迁移串联反应, 并进一步被氧化, 得到了二萜(-)-pal- lavicinin (153)和(+)-neopallavicinin (154). 随后, 他们通过引入C(2)双键改变了aldol反应位点——在酸性下, 二酮155发生C(4)-C(8)跨环aldol-O-Michael串联反应, 得到了二萜(+)-pallambin C (157)和(+)-pallambin D (158). 其中, 跨环aldol反应在温和弱酸磷酸二苯酯条件下收率较高; 然后, 在一锅中直接加入对甲苯磺酸(PTSA)可引发O-Michael反应. 该串联反应亦可直接由对甲苯磺酸引发, 但收率较低. 作者将此归咎于原料155对强酸的敏感性. (+)-pallambin C (157)和(+)- pallambin D (158)在光照下被进一步转化为了其它四种二萜(+)-pallambin A (159)、(+)-pallambin B (160)、(+)-pallaviambin A (161)和(+)-pallaviambin B (162). 该工作体现了仿生合成策略在天然产物全合成中的巨大潜力.
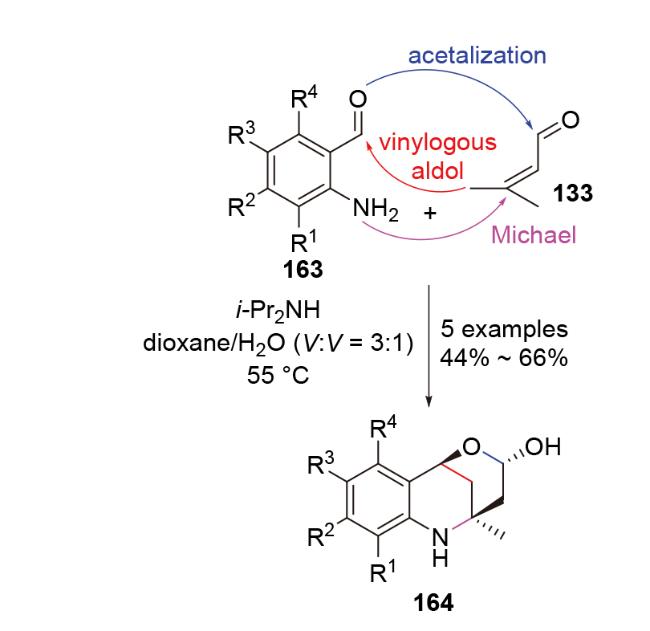
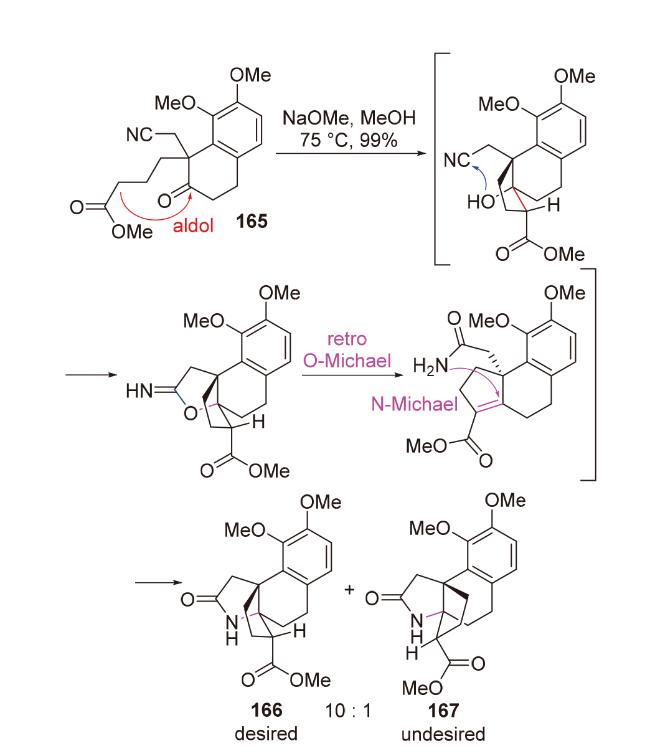
3.2 Aldol-N-Michael串联反应
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 总结与展望
综上所述, 按照Michael/aldol反应发生的先后顺序和Michael加成中亲核试剂的不同原子类型进行分类, 以四十余篇已报道的天然产物合成研究为例, 介绍了九类Michael/aldol串联环化反应在天然产物合成中的应用. 一方面, 该反应在构建复杂天然产物分子的核心骨架中显示出巨大潜力; 另一方面, 该反应在复杂分子的后期修饰方面亦具有显著优势, 为天然产物的简洁合成提供了多样化的途径和灵活的操作空间. 虽然, 该类反应在合成中已被广泛应用, 成果斐然, 但在底物的适用性、官能团兼容性以及反应的选择性等方面仍旧存在一定局限. 21世纪以来新发展的有机小分子催化方法[64-67]在该反应中应用颇多, 并以优良的反应选择性著称, 但依然具有收率不高、反应时间长及转化不完全等效率方面的缺陷. 未来, 开发条件更为温和、转化效率更高及操作更为简单的合成方法学, 仍将是该反应发展历程中兼具挑战和应用价值的重要课题.
(Zhao, C.)