

1 引言
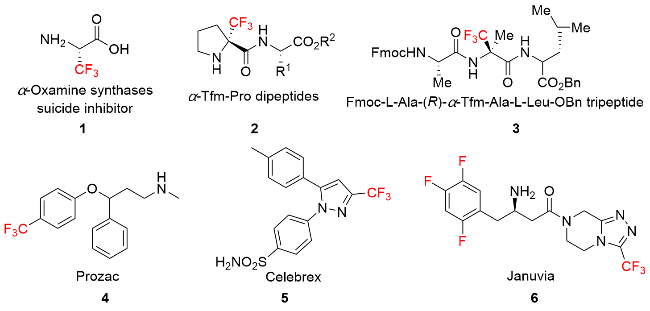
由于氟原子电负性强及原子半径小的特点, 在有机分子中引入氟原子能使化合物的亲脂性、与蛋白质间的相互作用及代谢的稳定性增加, 从而使药物的生物活性显著增强. 近年来, 含氟有机化合物在生物医学研究、治疗和诊断方面都有着广泛的应用, 使有机氟化学处于发展的黄金时期. 其中, 含三氟甲基的化合物是一类独特的化合物, 广泛用于医药、农药和材料等领域[1], 例如含三氟甲基的氮杂环药物Celebrex是治疗关节炎和骨质疏松所需要的药物, 对运动员的健康修复具有重要意义; 含三氟甲基的三氮唑Januvia, 配合饮食控制和运动, 对于改善II型糖尿病患者的血糖控制有良好效果[2](图1). 因此, 发展简洁高效的—CF3键形成及转化的新方法极其重要.
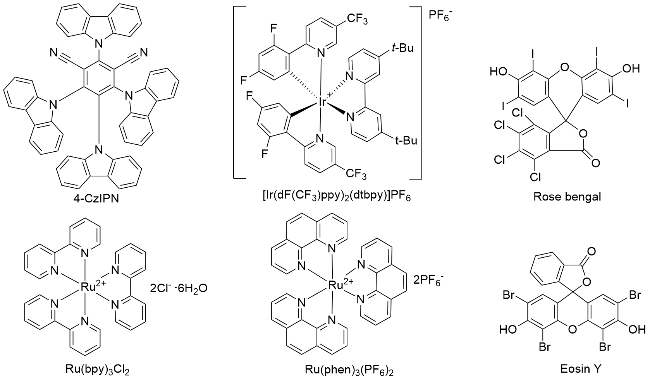
可见光具有资源充沛、廉价清洁和绿色无污染等特点, 如果能够直接利用可见光作为可持续能源实现有机化学的转化, 无论从经济还是环境的角度考虑都具有重要的研究意义. 近年来, 快速发展的可见光催化技术利用清洁的光能可以在温和的条件下引发自由基反应, 为官能化合物的合成提供了一种温和、有效的绿色合成方法[3]. 含三氟甲基的化合物具有独特的理化性质, 而含三氟甲基化合物的合成与转化也是多种多样的[4], α-三氟甲基烯烃作为一类制备含氟化合物的通用结构单元, 其选择性碳氟键活化引起了化学家的广泛的关注. 目前, 已有超过40例围绕三氟甲基烯烃多样性转化的研究报道, 有通过三氟甲基烯烃不脱氟方面来综述相关文献的, 有通过不同自由基使三氟甲基基团C—F键官能化的角度来综述相关文献的, 也有通过光催化促使三氟甲基烯烃脱氟方面来综述相关文献的[5], 而通过三氟甲基烯烃不同类型的转化来综述相关文献的相对较少. 在此, 本文将近年来在光化学条件下三氟甲基烯烃的多样性转化, 按照光催化下α-三氟甲基烯烃基于其双键的转化、基于自由基/极性交叉的SN2型转化进行综述(图2), 并且对本文涉及的光催化剂进行了绘图描述(图3), 部分的反应机理进行了讨论.
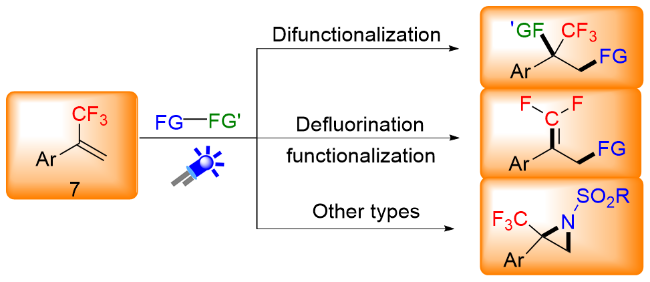
2 三氟甲基烯烃基于其双键的转化
2.1 三氟甲基烯烃的双官能团化
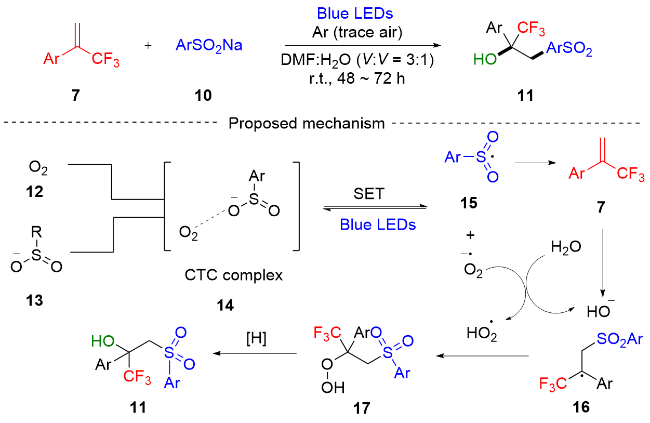
2021年, 阳华课题组[7]报道了光催化条件下α-三氟甲基苯乙烯与亚磺酸钠的磺酰基化-羟基化反应, 以中等至优良的产率得到了一系列α-三氟甲基-β-磺酰基叔醇化合物(Scheme 2). 该策略是通过氧气和亚磺酸盐之间的电荷转移复合物(CTC)机制实现的, 具有底物范围广泛和官能团兼容性好的优点. 课题组通过滴定实验和紫外吸收光谱实验提出了一个可能的反应机制. 在蓝色LED照射激发下, CTC复合物14内发生了单电子转移过程(SET), 产生磺酰基自由基15和超氧阴离子自由基. 磺酰基自由基和α-三氟甲基烯烃7发生自由基加成反应, 得到相应的自由基中间体16. 超氧阴离子自由基与水反应生成氢氧化物, 自由基中间体16和超氧阴离子自由基的自由基交叉偶联生成过氧化物中间体17. 最后, 可能通过芳基亚磺酸钠还原产生最终产物11.
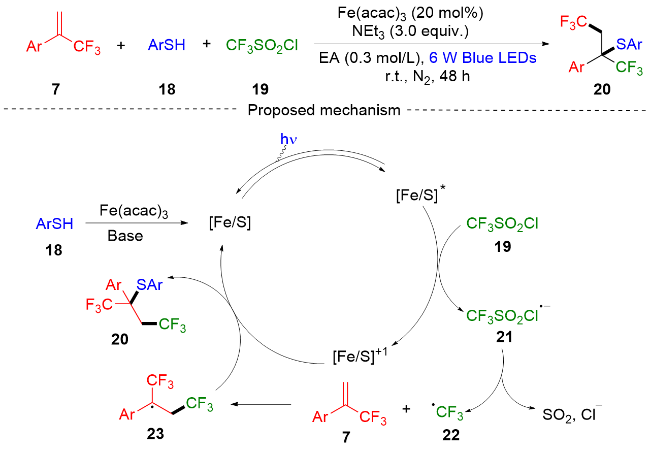
2022年, 李亚辉课题组[8]报道了一种在可见光的条件下α-三氟甲基烯烃的三氟甲基化-硫化反应(Scheme 3). 该反应以可见光为驱动力, 铁盐为催化剂, 硫醇为反应配体, 制备了一系列骨架新颖的α,β-双三氟甲基硫醇化合物. 生物活性化学物质也可以通过该方案提供所需的产品, 表明该合成方案可用于药物发现的后期修饰. 此外, 作者还在克级规模上开展了连续流合成, 表明了这些反应在工业生产中的应用前景. 课题组通过自由基抑制实验和紫外吸收光谱实验提出了一个可能的反应机制. 首先, 硫醇18和铁盐形成Fe/S络合物. 在可见光的照射下Fe/S络合物形成激发态Fe/S*, 激发态Fe/S*和三氟甲磺酰氯19发生反应得到高价态的Fe/S络合物和三氟甲磺酰氯自由基阴离子21. 然后, 三氟甲磺酰氯自由基阴离子分解为三氟甲基自由基22、二氧化硫、氯离子. 三氟甲基自由基对α-三氟甲基苯乙烯7进行自由基加成得到α-三氟甲基自由基中间体23, 接着和高价态的Fe/S络合物反应得到最终的三氟甲基化-硫化产物20, 并且Fe/S络合物进入下一轮催化循环.
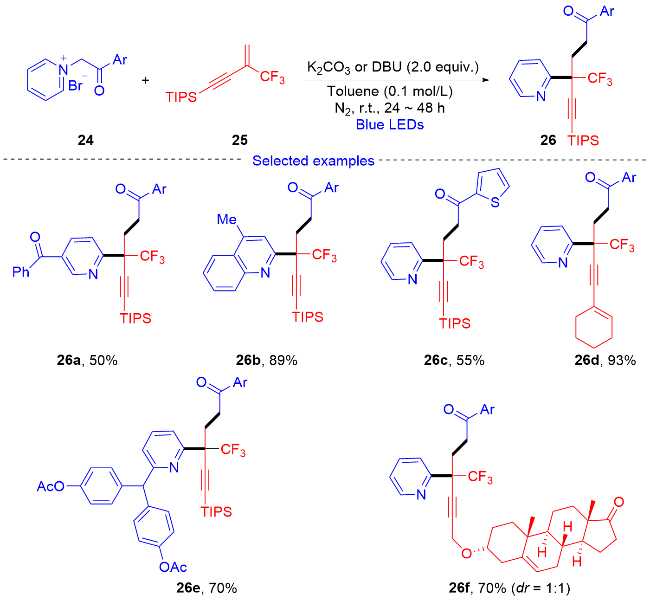
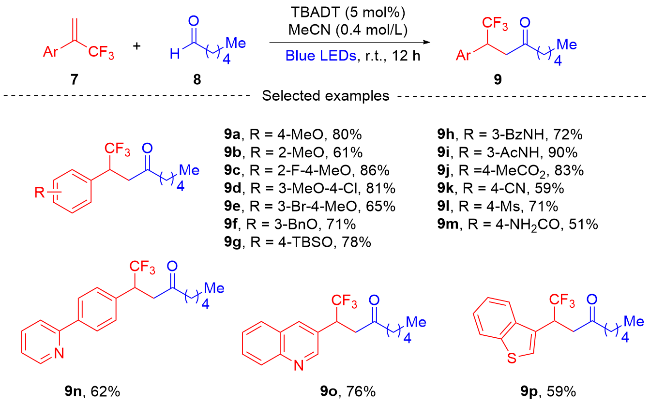
2.2 三氟甲基烯烃的环化
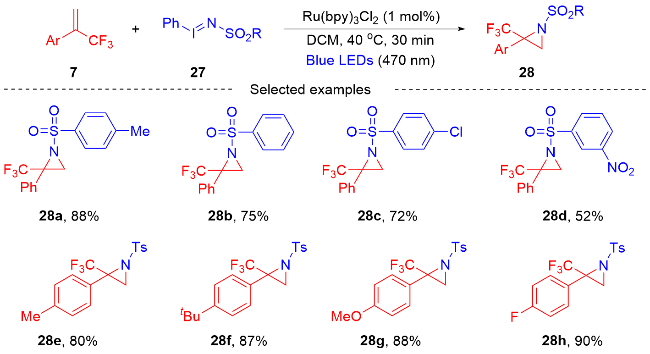
2.3 三氟甲基烯烃的可控不脱氟或脱氟
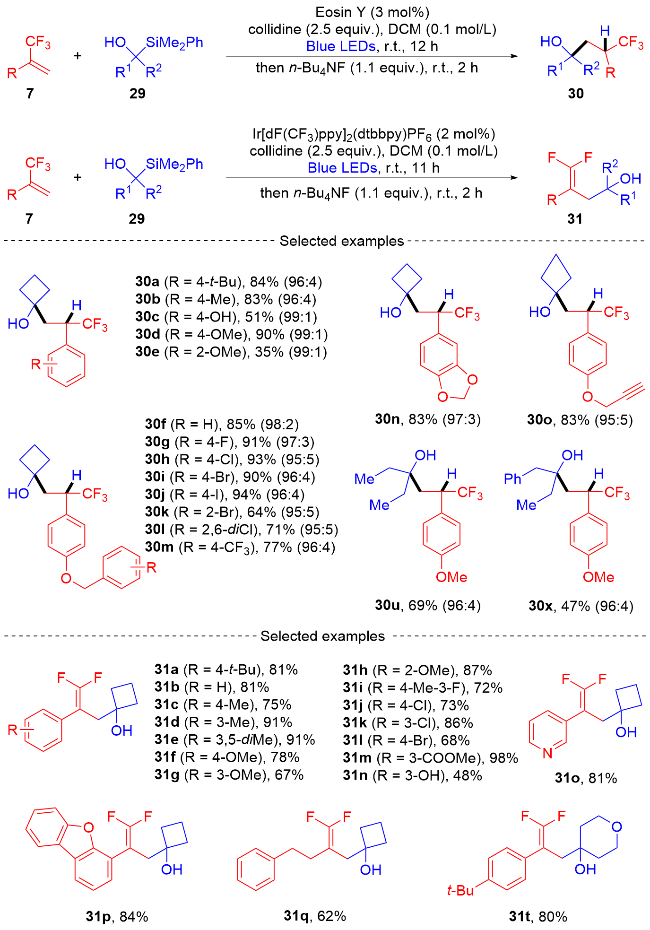
2024年, 傅尧课题组[12]介绍了一种通过电子给-受体(EDA)复合物策略进行的光诱导还原烷基化反应. 该方法通过EDA复合物策略实现了α-三氟甲基苯乙烯与N-羟基邻苯二甲酰亚胺酯之间的歧化脱羧反应和还原脱羧反应(Scheme 7). 该方法可以简单、高效地制备各种三氟甲基烷烃和二氟烯烃化合物, 具有高化学选择性和优异的官能团耐受性等优点. 初步机理研究表明, 烷基自由基由EDA复合物生成, 并与α-三氟甲基苯乙烯发生自由基加成反应, 生成α-三氟甲基苄基自由基、PPh3可作为还原剂, 在还原脱氟烷基化过程中使α-三氟甲基自由基生成苄基阴离子. 无过渡金属的温和反应条件、复杂分子的后期官能化和克级规模的合成以及连续流方式的成功都凸显了其合成潜力.
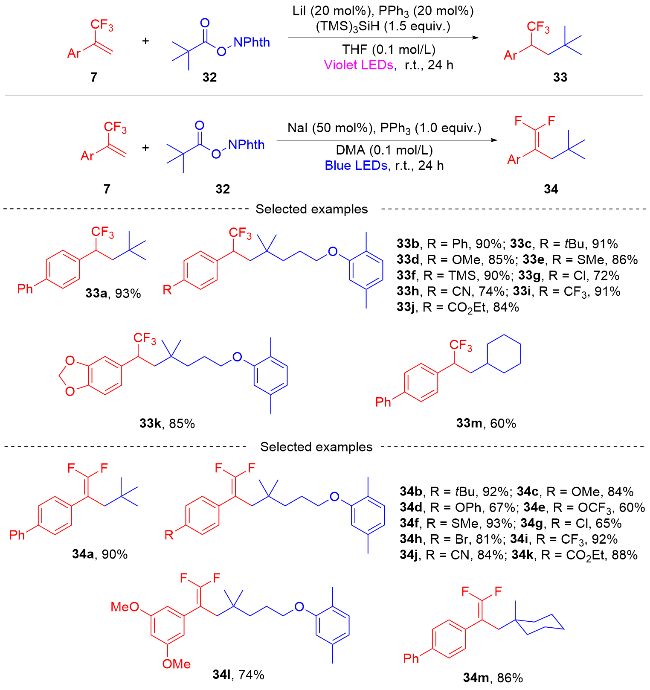
3 三氟甲基烯烃基于自由基/极性交叉的SN2型转化
3.1 三氟甲基烯烃转化为偕二氟烯烃
偕二氟烯烃类化合物在药物化学、材料化学和有机合成化学中具有广泛而重要的用途. 在小分子药物中引入偕二氟基团后, 分子脂溶性增强可促进其代谢稳定性, 进而提高药物的渗透性, 促进药物分子的吸收、分布及其与生物靶点的相互作用. 将廉价、易得的三氟甲基转化为偕二氟基团, 在有机合成中具有重要意义. 但是选择性断裂C—F键面临巨大的挑战, 因为三氟甲基中C—F键断裂的解离能远高于产物偕二氟基团中C—F键解离能, 难以控制C—F键的选择性断裂, 极易发生过度脱氟[13]. 相对于传统热反应, 在可见光促进下, 利用光催化剂敏化底物, 创造一条经过激发态的新反应途径, 可以更加高效、高选择性地实现C—F断裂.
2016年, 周磊课题组[14]描述了一种光催化下α-三氟甲基烯烃与α-酮酸和α-氨基酸的脱羧和脱氟反应, 以优良的产率得到了一系列二氟烯丙基酮和二氟烯丙基胺化合物(Scheme 8). 首先, 他们以苯甲酰甲酸和α-三氟甲基苯乙烯进行了反应条件筛选, 发现当使用Li2CO3作为碱时副产物可以被抑制. 当改变碱时, 产物的产率降低, 表明了锂离子可以促进氟原子的离去能力, 从而有利于主产物的生成. 他们进行了对照试验表明由于氢氧化锂的碱性原因, 副产物中的三氟甲基基团并不会发生α位的氢原子去质子化, 因此从副产物中转化得到主产物是不太可能的. 在确定了最佳的反应条件后, 该合成方法以中等至良好的产率得到了相应的偕二氟烯丙基酮和偕二氟烯丙基胺化合物.
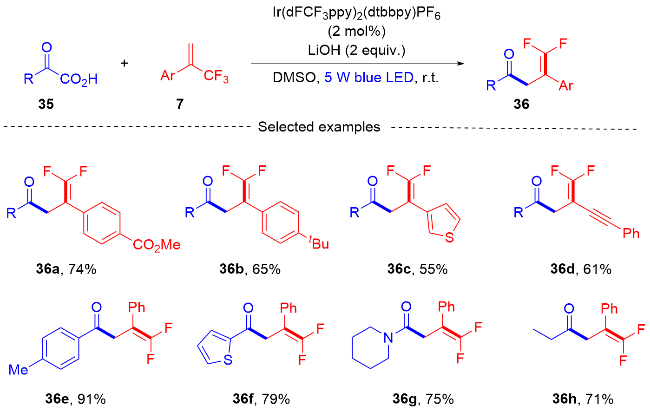
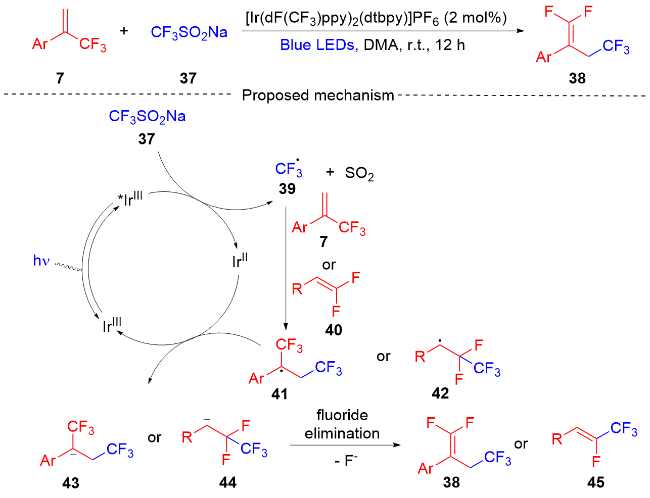
2018年, 沈志良及其同事[15]报道了一种光氧化还原催化α-三氟甲基烯烃和偕二氟烯烃的脱氟三氟甲基化反应(Scheme 9). 该反应以CF3SO2Na作为三氟甲基源, 通过光氧化还原催化, 获得了具有立体选择性的多氟有机分子. 课题组对此反应进行了对照试验, 表明了光和光催化剂在该反应中是必不可少的. 该反应通过Ir光氧化还原催化的C—F键断裂, 开发了α-三氟甲基烯烃和偕二氟烯烃的脱氟三氟甲基化反应, 提供了一种方便有效的方法来构建取代的二氟烯烃. 此外, 课题组在自由基捕获实验的基础上, 提出了一个可能的反应机制. 在可见光的照射下, 光催化剂IrⅢ变成激发态*IrⅢ, 三氟甲基源CF3SO2Na和激发态的光催化剂*IrⅢ发生单电子转移(SET)过程. 三氟甲基源37被氧化形成三氟甲基自由基39和二氧化硫, 三氟甲基自由基和三氟甲基烯烃7或偕二氟烯烃40进行自由基加成, 得到双三氟甲基自由基中间体41或单三氟甲基自由基中间体42. 其随后和光催化剂IrⅡ发生单电子转移(SET)过程光催化剂获得再生, 双三氟甲基自由基中间体41和单三氟甲基自由基中间体42被还原为碳负离子中间体43或44. 碳负离子中间体容易发生β-氟化物的消除, 最终得到产物38或45.
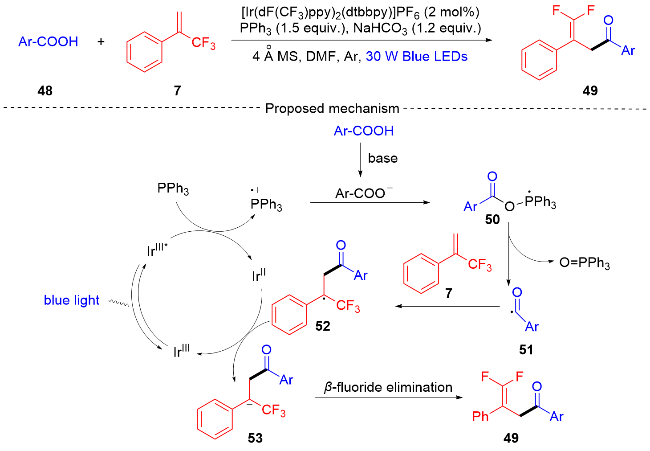
2020年, 汪清民课题组[17]描述了一种高效、实用的光催化脱氧或脱氟合成γ,γ-二氟烯丙基酮的方法(Scheme 11), 该方案不仅适用于芳香族羧酸和苯乙酸型脂肪族羧酸, 而且适用于芳香族杂环羧酸. 例如, 呋喃、噻吩、吡咯和吲哚等, 优良的官能团耐受性和温和的反应条件证明了该合成方案的潜力. 课题组通过自由基捕获实验和相关文献报道[18], 提出了二氟烯丙基酮合成反应可能的反应机制. 首先, 在蓝光的照射下光催化剂IrⅢ被激发成IrⅢ*发生单电子转移过程[19], 激发态IrⅢ*被PPh3淬灭[20]形成三苯基膦自由基阳离子和IrⅡ. 在碳酸氢钠的存在下, 自由基阳离子和羧酸底物反应生成膦酰基自由基中间体50. 该自由基中间体经过β-选择性C—O键断裂形成三苯基氧化膦和酰基自由基51, 酰基自由基和α-三氟甲基烯烃7发生自由基加成反应生成自由基中间体52, 自由基中间体被IrⅡ物质还原成碳负离子中间体53, 光催化剂得到再生从而完成光催化循环. 最后, 碳负离子中间体发生β-氟化物的消除反应得到γ,γ-二氟烯丙基酮产物49.
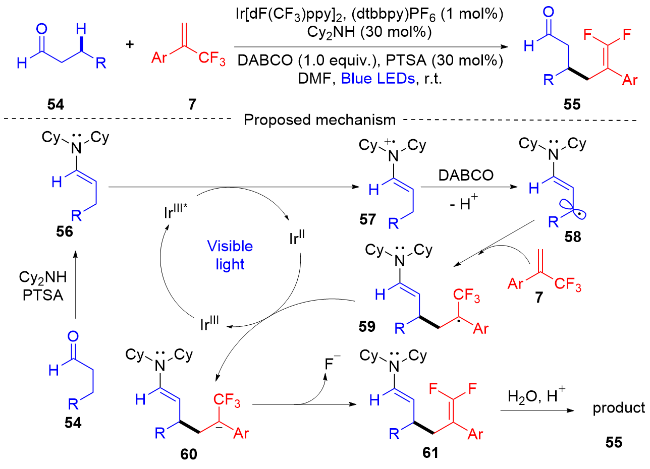
2020年, 周磊课题组[21]描述了一种可见光催化的醛和环酮β-C—H偕二氟烯丙基化反应(Scheme 12). 该方案在光氧化还原催化和有机催化的协同组合下, α-三氟甲基烯烃通过C—F键的断裂被用作偕二氟烯丙基化试剂. 酸催化剂、有机催化剂和光催化剂的协同组合对于这种β-C—H官能化过程至关重要. 所得的羰基官能化偕二氟烯烃可用作许多转化的通用结构单元, 这一工作是对目前广泛研究的偕二氟烯烃的一个很好的补充. 此外, 课题组提出了一个可能的反应机制: 在酸催化剂的存在下, 醛54与Cy2NH缩合形成烯胺55, 其被激发的光催化剂IrⅢ*单电子还原, 得到自由基阳离子中间体57[22]. 自由基阳离子中间体的酸性β-C—H被1,4-二氮杂二环[2.2.2]辛烷(DABCO)去质子化, 形成亲核β-烯胺基自由基中间体58. β-烯胺基自由基中间体很容易与亲电烯烃自由基受体7偶联, 形成所需要的C—C键, 同时产生三氟甲基自由基中间体59. 三氟甲基自由基中间体和光催化剂IrⅡ发生单电子转移(SET)过程, 三氟甲基自由基中间体被还原为三氟甲基碳负离子中间体60, 同时再生处于基态的光催化剂IrⅢ. 最后, 从三氟甲基碳负离子中间体中消除β氟化物, 水解得到所需的偕二氟烯丙基化产物55.
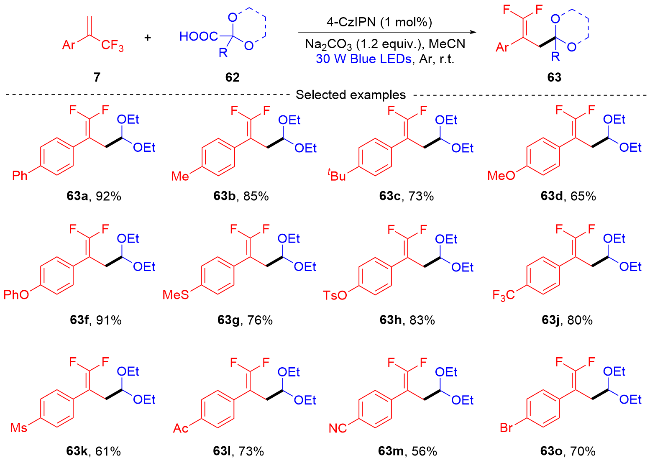
2021年, 潘菲课题组[26]报道了一种通过光氧化还原催化的自由基交叉偶联反应(Scheme 14), 开发了内酰胺取代的偕二氟烯烃的合成. 该反应是一种简洁的制备内酰胺、氨基甲酸酯和脲取代偕二氟烯烃的方法, 是通过光氧化还原催化、Bronsted辅助的分子内5-exo-trig环化/分子间自由基加成/β-氟消除反应等过程合成的内酰胺取代偕二氟烯烃. 课题组根据自由基捕获实验和自由基钟实验以及相关文献报道[27]的基础上提出了一个可能的反应机制. 首先, 在Bronsted碱的帮助下, 酰胺64发生单电子转移(SET)过程以产生酰胺基自由基66. 然后发生自由基的未活化烯烃分子内加成, 得到带有内酰胺的烷基自由基物质67. 烷基自由基物质67对α-三氟甲基烯烃7进行自由基加成, 形成自由基中间体68. 随后, 自由基中间体68通过IrⅡ的单电子还原得到碳负离子中间体69, 并且再生光催化剂. 最后, 碳负离子中间体69发生β-氟消除, 生成内酰胺修饰的偕二氟烯烃65, 并完成了催化循环.
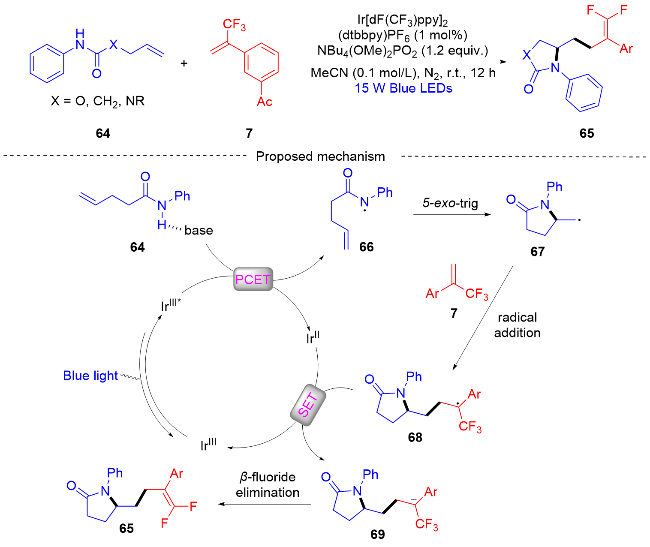
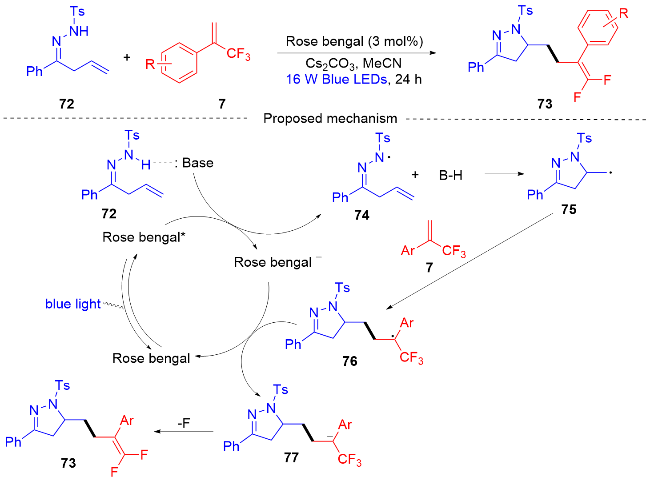
2021年, 周明东课题组[29]报道了一种可见光促进的N-自由基介导的β,γ-不饱和腙和α-三氟甲基烯烃的自由基串联环化/脱氟烷基化反应(Scheme 16). 该方案提供了一种在中性氧化还原、无金属和温和条件下, 以中等至优异的产率合成各种二氢吡唑稠合偕二氟烯烃化合物. 这种无金属参与的反应, 对于敏感官能团以及结构复杂的有机化合物表现出良好的取代基兼容性. 通过自由基抑制实验和荧光淬灭实验以及相关文献报道[30-31], 作者提出了一个可能的反应机制. 首先, 在可见光的照射下形成激发态Rose bengal*, 其可以氧化不饱和腙72, 在碱的帮助下形成N-中心腙基自由基74. 接下来, N-中心腙基自由基加成到末端烯烃上形成碳中心自由基75, 碳中心自由基对α-三氟甲基烯烃7进行自由基加成反应, 得到碳中心自由基中间体76. 在此之后, 碳中心自由基中间体76被Rose Bengal–还原为碳负离子中间体77, 发生了一个单电子转移(SET)过程. 最后, 从碳负离子中间体中消除氟原子, 得到了最终的产物偕二氟烯烃化合物73.
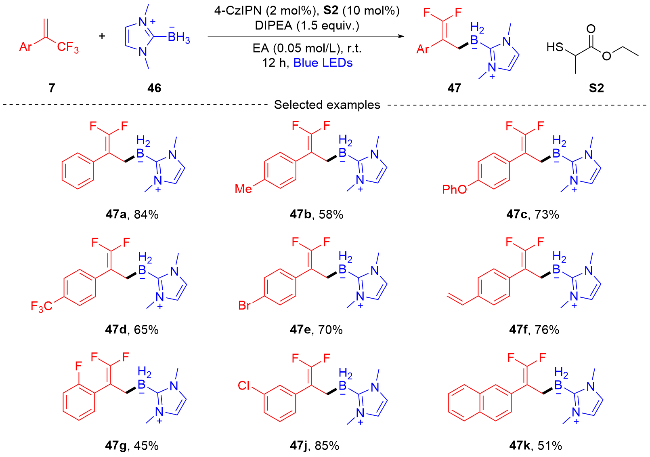
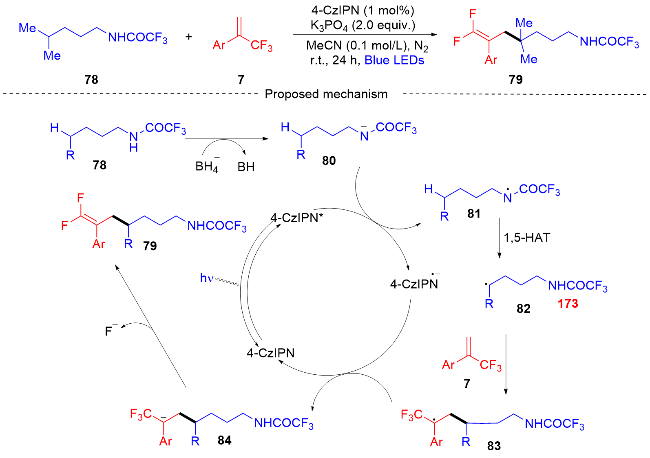
2021年, 潘菲课题组[32]报道了一种通过光催化产生酰胺基自由基对酰胺中未活化的C—H键进行远程位点选择性官能化的反应(Scheme 17), 以形成具有三氟甲基取代的偕二氟烯烃化合物. 位点选择性是由酰胺的1,5-氢原子迁移(HAT)过程控制, 这种光催化转化有利于形成二级、三级和四级碳中心, 酰胺基自由基可以有效的原位产生. 1,5-氢原子迁移(HAT)过程可以高选择性地形成与碳原子进行反应的自由基物种. 此外, 课题组在自由基捕获实验和自由基抑制实验以及相关文献报道[33-34]的基础上提出了一个可能的反应机制. 最初,在可见光的激发下光催化剂4-CzIPN成为了激发态4-CzIPN*, 原始底物78被拔氢形成氮负离子中间体80, 氮负离子中间体和激发态的4-CzIPN*发生单电子转移(SET)过程. 氮负离子中间体形成酰胺基自由基中间体81, 接着从氮到碳的1,5-氢原子迁移(HAT)过程产生碳中心自由基中间体82. 碳中心自由基中间体和三氟甲基烯烃7发生自由基加成反应, 得到自由基中间体83, 自由基中间体83被4-CzIPN自由基阴离子还原生成碳负离子中间体84, 并再生光催化剂4-CzIPN. 最后, 在碳负离子中间体中消除β-氟化物产生所需的偕二氟烯丙基化产物79.
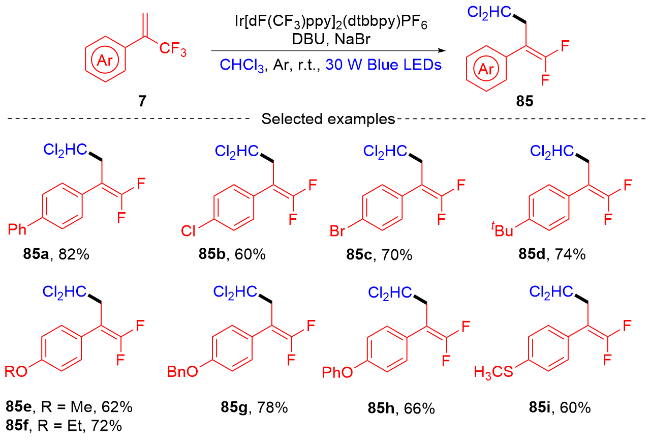

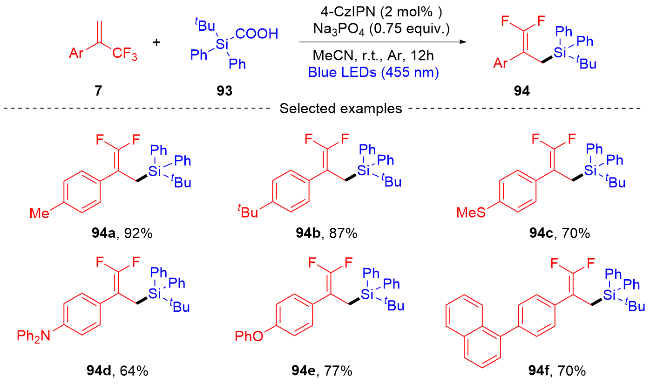
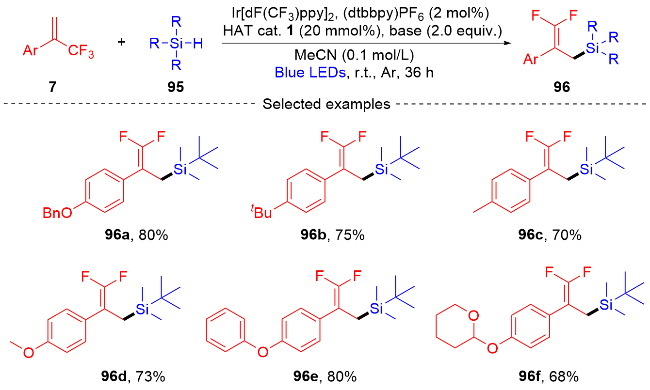
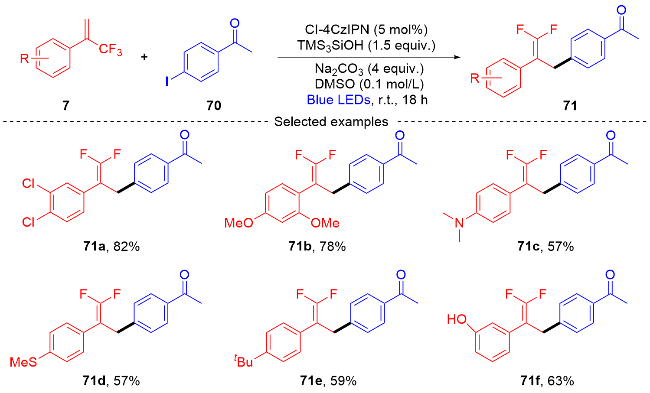
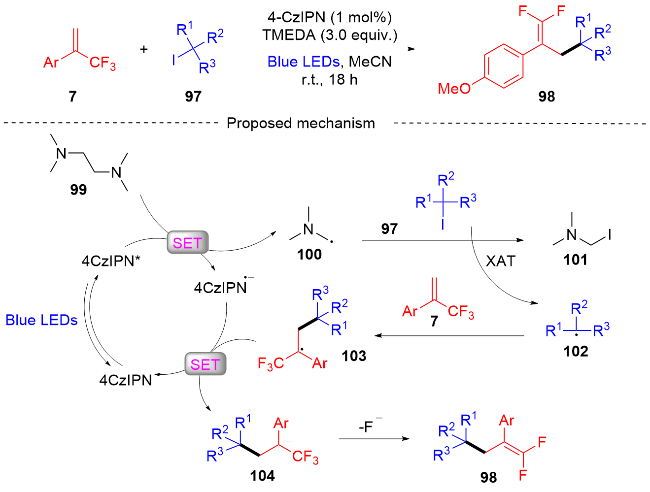
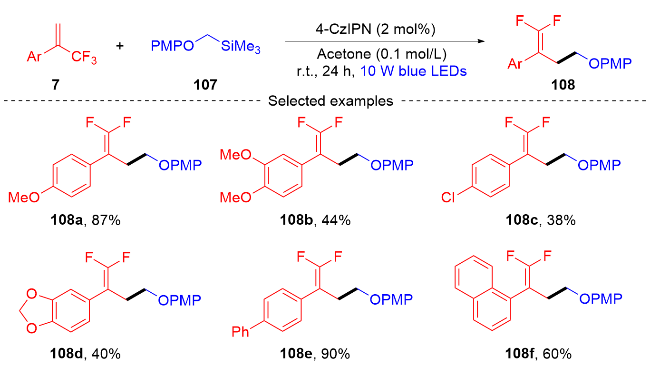
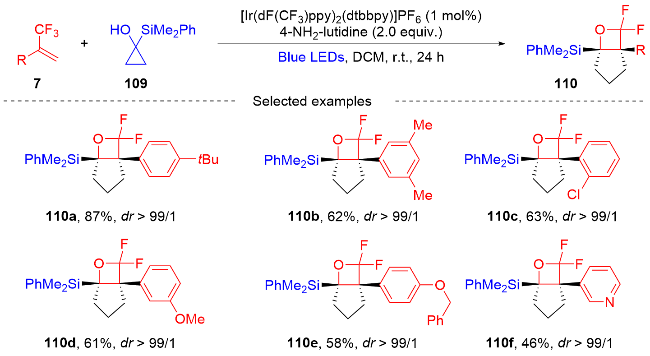
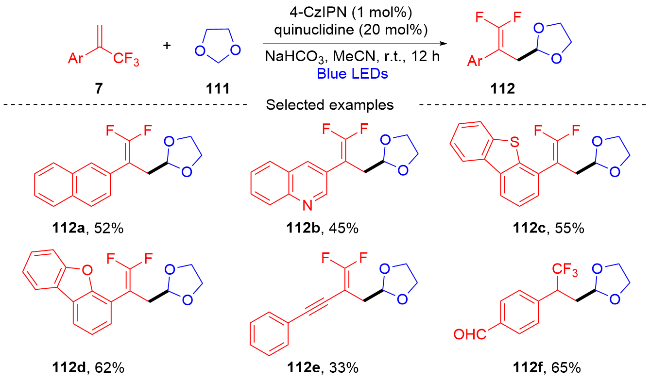
2022年, 张振明课题组[41]报道了一种可见光催化的α-三氟甲基烯烃与碘代烷的偕二氟烯丙基化反应 (Scheme 24), 通过C—F键的断裂合成偕二氟衍生物. 该反应以4-CzIPN作为光催化剂, 并采用常见的四甲基乙二胺作为卤素原子转移自由基活化剂, 提供了一种具有良好官能团耐受性的偕二氟烯烃化合物路线. 此外, 课题组在自由基捕获实验的基础上, 提出了一个可能的反应机制. 首先, 在蓝光的激发下光催化剂4-CzIPN变成激发态4-CzIPN*, 其通过单电子转移(SET)过程和C—C键的断裂过程与TMEDA反应形成α-氨基烷基自由基100和4-CzIPN自由基阴离子. 然后, α-氨基烷基自由基和烷基碘97发生卤素原子转移(XAT)过程生成烷基自由基102. 随后, 烷基自由基102和α-三氟甲基烯烃7发生自由基加成反应生成自由基中间体103, 自由基中间体103被4-CzIPN自由基阴离子还原为碳负离子中间体104, 光催化剂4-CzIPN得到再生. 最后, 碳负离子中间体发生β-F的消除反应, 得到最终的偕二氟烯烃产物98.
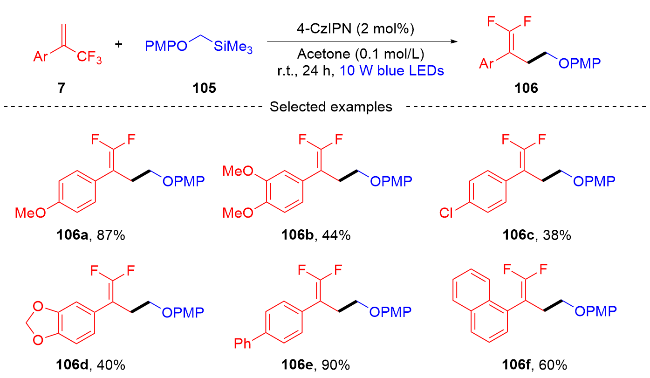
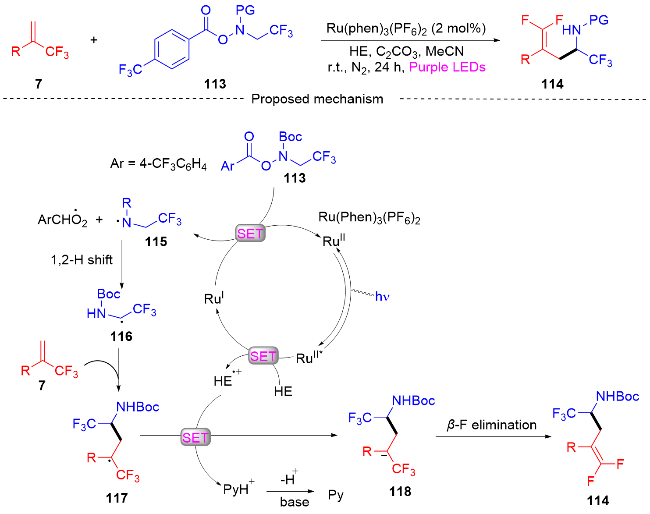
2023年, 陈芬儿课题组[46]介绍了一种可见光诱导的烷基化反应(Scheme 29). 该反应通过光氧化还原催化α-三氟甲基烯烃与N-三氟乙基羟胺的自由基加成/脱氟烷基化偶联等过程合成偕二氟烯丙基化α-三氟甲基胺. 此方案通过原位生成的N-三氟乙基自由基的1,2-H迁移实现自由基的加成反应, 合成了一系列偕二氟烯丙基化的α-三氟甲基胺化合物. 与传统的亚胺转化法合成α-三氟甲基胺不同, 该方法提供了一种可供选择的高效单电子合成方法, 可用于更复杂底物的后期功能化. 此外, 该课题组在自由基捕获实验和荧光淬灭实验及相关文献的基础上, 提出了一条可能的合成机理. 最初, 在可见光的照射下光催化剂RuⅡ被激发成RuⅡ*, 其接下来经历与2,6-二甲基-1,4-二氢-3,5-吡啶二羧酸酯(HE)的单电子转移(SET)过程以产生还原的RuⅠ物质和HE的自由基阳离子. 三氟乙基羟胺试剂113和光催化剂RuⅠ发生单电子转移(SET)过程产生N-中心自由基115和再生的光催化剂RuⅡ. 随后, N-中心自由基发生1,2-H迁移形成更稳定的自由基116. 自由基116和α-三氟甲基烯烃7发生自由基的加成反应得到三氟甲基自由基中间体117, 自由基中间体117被HE自由基阳离子还原生成碳负离子中间体118, 碳负离子中间体接着发生β-氟消除反应得到所需的偕二氟烯丙基化α-三氟甲基胺类化合物114.
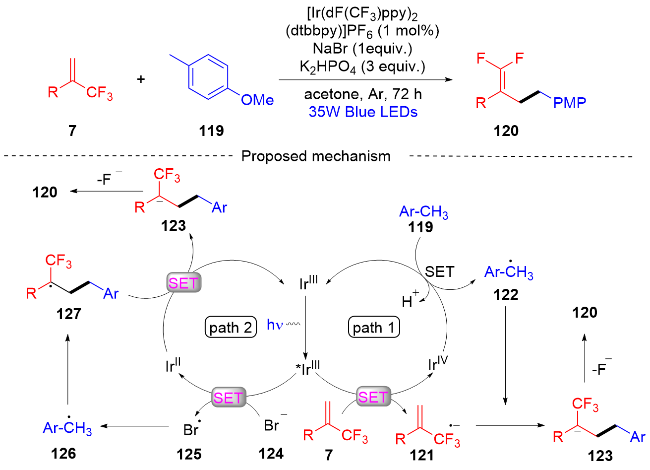
2023年, 姬小趁课题组[47]报道了一种三氟甲基烯烃与烷基芳烃的光氧化还原脱氟苄基化反应 (Scheme 30). 该反应在温和的反应条件下, 提供了一系列在结构上有价值的苄基偕二氟烯烃化合物的方案. 根据初步的机理研究表明, 反应途径和反应速率是苄基自由基形成的重要因素. 在自由基捕获实验、自由基钟实验、循环伏安实验的基础上, 作者提出了光催化三氟甲基烯烃与烷基芳烃的脱氟苄基化反应可能的机理. 首先, 光催化剂IrⅢ在蓝色LED照射下生成激发态*IrⅢ, 其与α-三氟甲基烯烃7发生单电子转移(SET)过程, 得到相应的自由基阴离子121和被氧化的IrⅣ. 烷基苯119被IrⅣ氧化, 随后去质子化并使光催化剂得到再生, 烷基苯119则生成苄基自由基122, 苄基自由基和自由基阴离子121发生自由基加成反应, 然后脱除乙酰基得到偕二氟烯烃产物120(路径1). 或者, 在溴化物添加剂的存在下, 可以通过激发的光催化剂还原淬灭循环形成溴自由基, 随后夺取烷基苯的氢原子以产生关键的苄基自由基(路径2).
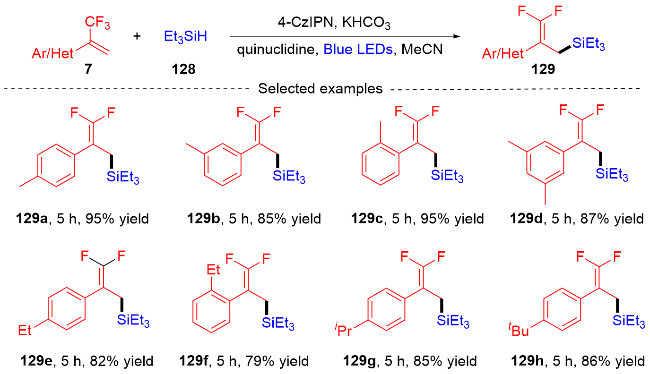
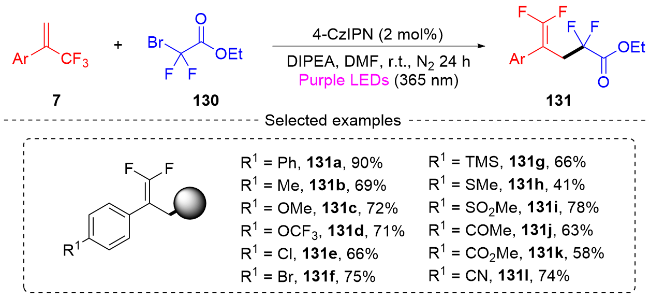
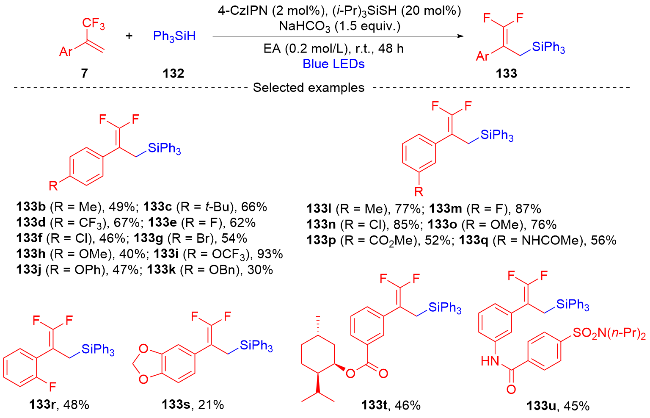
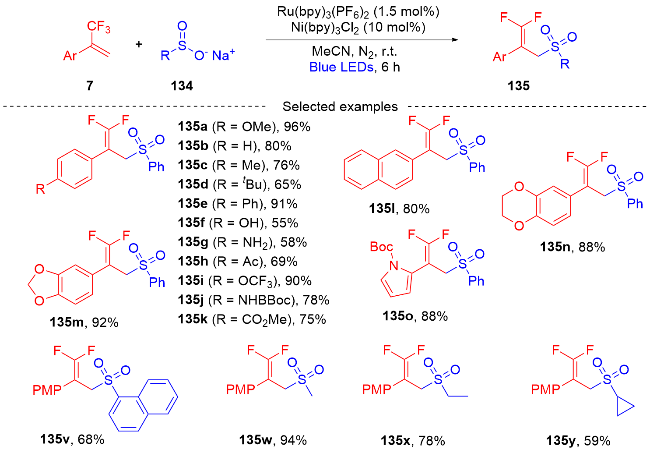
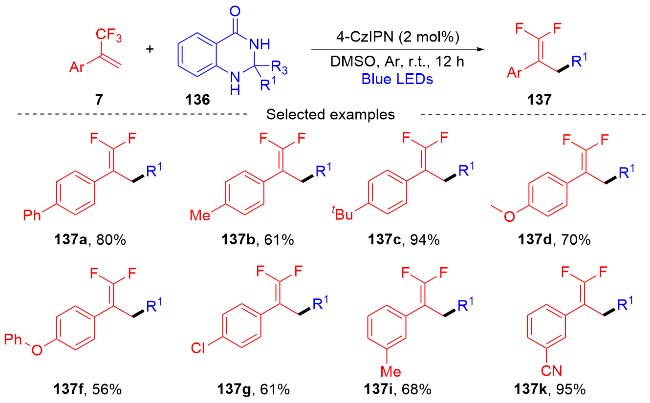
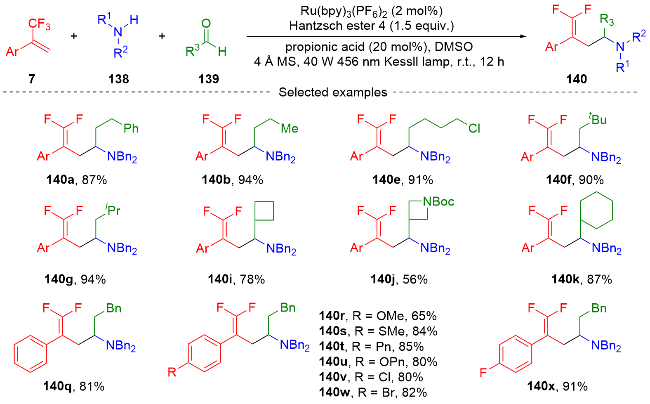
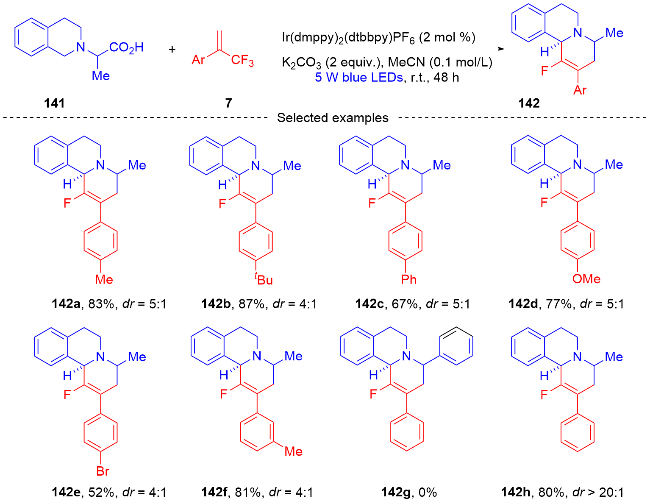
3.2 三氟甲基烯烃转化为单氟杂环
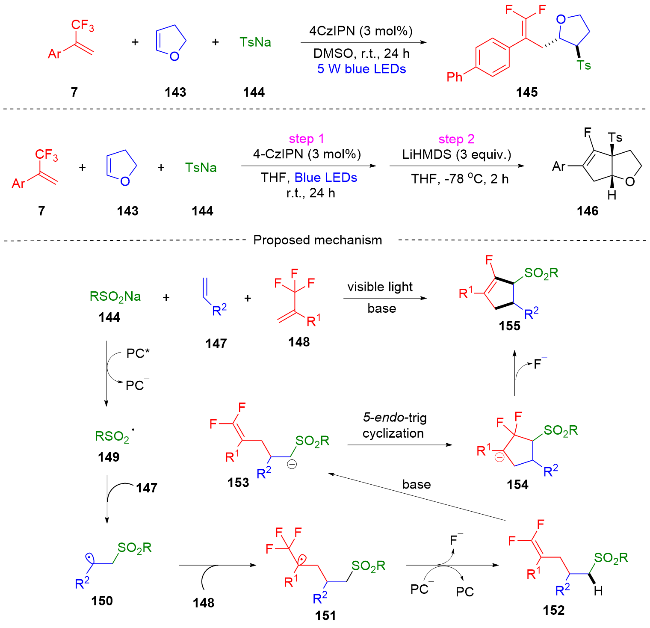
2022年, 周磊课题组[55]报道了一种在光催化下α-三氟甲基烯烃、烯烃和亚磺酸钠脱氟三组分合成单氟环戊烯的反应(Scheme 38). 该反应中涉及的亚磺酸钠具有双重作用, 不仅作为引发两种不同烯烃的光催化脱氟偶联的自由基前体, 而且通过引入强吸电子磺酰基来增加α-C—H键的酸性. 由于磺酰基的强吸电子性质, 所得的偕二氟烯烃可以通过分子内碱介导的分子内环化(SNV)反应, 在一锅内转化为各种单氟环戊烯. 在这项工作中, 他们通过自由基捕获实验提出了一个可能的反应机制. 首先, 亚磺酸钠144和光催化剂发生单电子转移过程, 产生了磺酰基自由基149, 其经历与烯烃147的加成反应得到β-磺酰基烷基自由基150, 自由基150被α-三氟甲基烯烃148捕获, 生成三氟甲基碳自由基151. 然后进行SET还原和β-氟的消除得到偕二氟烯烃152, 由于缺电子的磺酰基增加了α-位碳氢键的酸性, 因此生成的偕二氟烯烃152分子内(SNV)反应可能会产生单氟环戊烯155.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论
本文对近年来在光化学条件下三氟甲基烯烃的多样性转化, 按照光催化下α-三氟甲基烯烃基于其双键的转化、基于自由基/极性交叉的SN2型转化, 进行了较为详细地阐述, 并对部分反应的底物范围和机理进行了讨论. 从以上论述可以看出, 在光化学条件下三氟甲基烯烃的多样性转化, 已经成为了有机化学家近年来研究的热点. 虽然在光化学条件下三氟甲基烯烃的转化研究已获得较大进展, 但多数情况下都是脱氟生成偕二氟烯烃类化合物; 此外, 通过三氟甲基烯烃的双官能团化途径实现季碳中心的构建策略相对较少, 并且有些季碳中心构建的方案还必须在过渡金属催化条件下进行, 反应存在局限性. 近年也有少数文献报道, 通过不同光敏剂的调控可以同时获得不脱氟产物和偕二氟烯烃产物. 因此, 在光化学条件下三氟甲基烯烃的双官能团化反应还有待发展, 开发更为绿色高效, 经济实用的双官能团化反应, 并将其应用于工业化生产与药物研发, 是后期值得关注的发展方向. 此外, 通过不同的调控手段同时获得多种含氟产物是一个值得关注的研究方向. 相信随着研究的不断深入, 未来在光化学条件下三氟甲基烯烃的转化应用研究一定会更加丰富.
(Lu, Y.)