

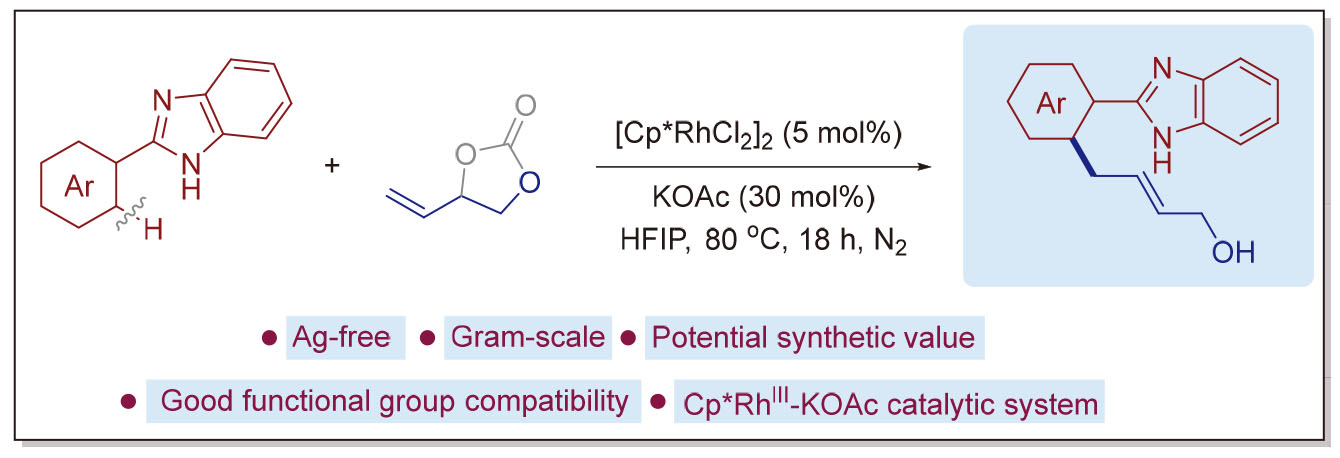
1 引言
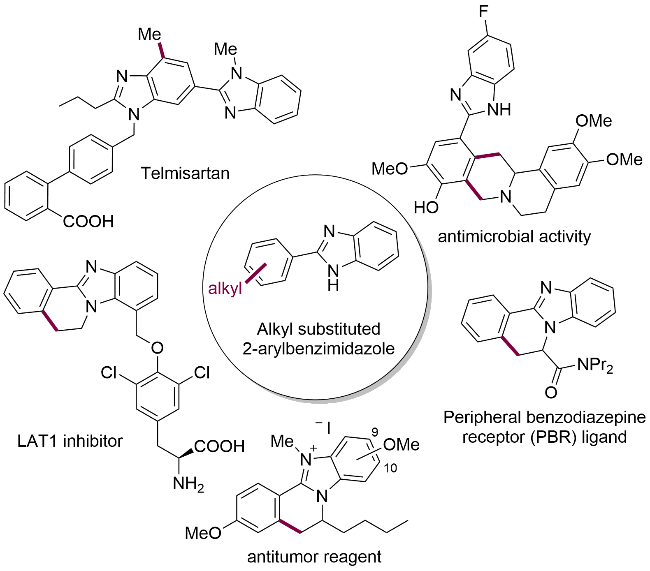
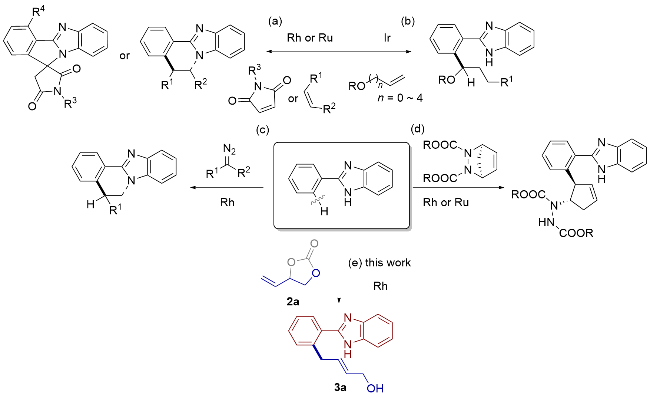
近年来, 2-芳基苯并咪唑类化合物得到了广泛的研究[9a-9b], 其中C—H烷基化反应已取得系列突破性进展. Sun课题组[9c-9d]报道了以苯乙烯或马来酰亚胺作为烷基化试剂的反应体系, 成功开发出苯并咪唑并环及螺环产物的高效合成策略(图2a). 然而值得注意的是, 仅当使用2-乙烯基吡啶作为底物时, 才能观察到未发生环化的直接烷基化产物. Nishimura课题组[10]则通过采用烯基醚作为底物, 建立了2-芳基苯并咪唑的对映选择性和支链选择性C—H烷基化反应体系(图2b). 2018年, 宋秋玲课题组报道了在Cp*RhIII催化下, 2-芳基苯并咪唑与α-重氮酮酯的C—H烷基化反应, 该反应可定向合成咪唑并[2,1-a]异喹啉类化合物[11]或直接烷基化的2-芳基苯并咪唑衍生物[12](图2c). 鉴于重氮化合物存在稳定性差、易爆性和毒性等明显缺陷, 开发易得且安全的烷基化试剂具有重要的意义. 烯丙基作为最重要的烷基片段之一, 其双键官能团可进一步转化为多种有用官能团[13]. 然而, 2-芳基苯并咪唑的C—H烯丙基化反应研究仍十分有限, 目前仅有Radhakrishnan课题组[14]报道的一例工作——在铑或钌催化下使用二氮杂双环烯烃作为烷基化试剂(图2d). 基于近期我们课题组[15]在2-芳基咪唑C—H官能团化的研究进展, 本研究首次报道了铑(III)催化的2-芳基苯并咪唑与商业可得的试剂4-乙烯基-1,3-二氧戊环-2-酮(2a) C—H烯丙基化反应(图2e).
2 结果与讨论
2.1 反应条件优化
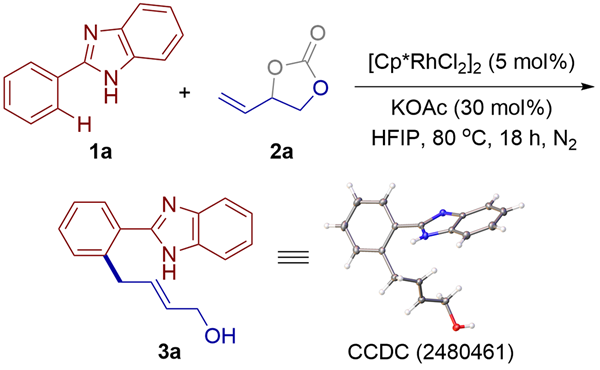
本研究以2-芳基苯并咪唑(1a)和4-乙烯基-1,3-二氧戊环-2-酮(2a)作为模版底物进行反应条件优化(表1). 经过系统的反应参数筛选, 确定了在氮气保护下, 以五甲基环戊二烯基二氯化铑二聚体([Cp*RhCl2]2, 5 mol%)作为催化剂、乙酸钾(KOAc, 30 mol%)作为添加剂、六氟异丙醇(HFIP)作为溶剂, 80 ℃反应18 h为最佳反应条件, 可获得61%收率的目标产物3a (表1, Entry 1). 值得注意的是, 尽管二氯(p-甲基异丙苯)钌(II)二聚体([Ru(p-cymene)Cl2]2)成本较低且在C—H活化中已有成功应用案例, 但在本研究体系中将其替代[Cp*RhCl2]2却未能得到目标产物3a (表1, Entry 2). 添加剂的筛选结果表明: 当使用HOAc或Na2CO3替代KOAc时, 反应完全不能进行; 而采用NaOtBu或K2CO3时, 产物3a的收率分别为35%和38% (表1, Entries 3~5). 溶剂筛选实验表明, 该转化反应对溶剂选择具有高度依赖性. 当采用甲苯(toluene)或二甲基亚砜(DMSO)时, 反应无法生成目标产物; 在乙腈(CH3CN)或四氢呋喃(THF)溶液中以11%的收率获得产物3a (表1, Entries 6~8), 这一结果充分证明溶剂六氟异丙醇(HFIP)在该转化反应中的不可替代性. 温度效应研究表明: 反应温度显著影响反应效率——当温度大幅降低时, 产物收率急剧下降(表1, Entry 9); 而升高反应温度却未能提高目标产物的收率(表1, Entry 10). 值得关注的是, 该反应在空气氛围中仍能顺利进行(表1, Entry 11). 对照实验证实, 反应体系的催化活性完全依赖于[Cp*RhCl2]2催化剂和KOAc添加剂——当去除其中任一组分时, 反应均被完全抑制(表1, Entry 12).
表1 反应条件的筛选aTable 1 Screening reaction conditons a |
| Entry | Deviation from optimal conditons | Yieldb/% |
|---|---|---|
| 1 | none | 61 |
| 2 | [Ru(p-cymene)Cl2]2 instead of [Cp*RhCl2]2 | 0 |
| 3 | HOAc or Na2CO3 instead of KOAc | 0 |
| 4 | NaOtBu instead of KOAc | 35 |
| 5 | K2CO3 instead of KOAc | 38 |
| 6 | Toluene or DMSO instead of HFIP | 0 |
| 7 | CH3CN instead of HFIP | 11 |
| 8 | THF instead of HFIP | 11 |
| 9 | 40 ℃ | 28 |
| 10 | 120 ℃ | 50 |
| 11 | under air | 50 |
| 12 | without [Cp*RhCl2]2 or KOAc | 0 |
a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), catalyst (5.0 mol%), KOAc (30 mol%), HFIP (1.5 mL), 18 h under N2; b Isolated yields. |
2.2 反应底物拓展
在确定最佳反应条件后, 本文系统考察了该转化反应的底物适用范围(表2). 虽然在该反应中获得的产率不是特别高, 但其成功实现了结构多样的2-芳基苯并咪唑类底物的有效转化, 表明其具有良好的兼容性. 首先, 带有甲基或叔丁基的2-芳基苯并咪唑在该体系中是可以生成目标产物3b和3c的. 值得注意的是, 芳环上的取代基(X=F, Cl, Br)不会干扰反应, 以中等但具有合成价值的收率(49%~64%)获得目标产物3d~3i. 出乎意料的是该反应能兼容带有游离羟基的2-芳基苯并咪唑并以41%的收率得到3j. 该反应可以兼容给电子基团如甲氧基的引入(3k和3l), 以可接受的收率获得相应产物. 缺电子的2-芳基苯并咪唑同样适用该体系(3m~3n). 最后, 其它芳香取代底物包括2-萘基以及杂芳环片段(如呋喃、苯并噻吩和吡啶)也都是该反应的合适底物(3o~3r).
表2 底物范围aTable 2 Substrate scope a |

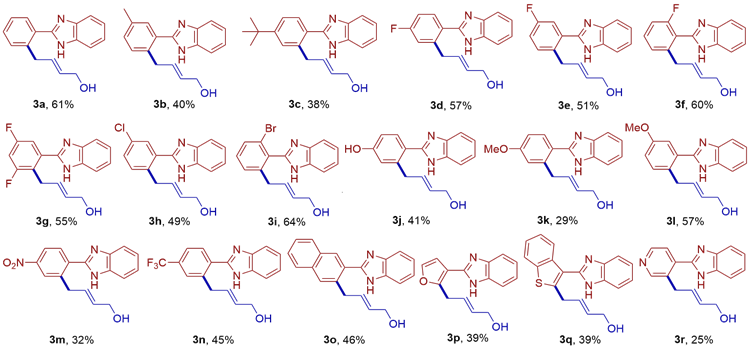 |
a Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), catalyst (5.0 mol%), KOAc (30 mol%), HFIP (1.5 mL), 18 h under N2; Isolated yields. |
2.3 模板化放大反应及其转化
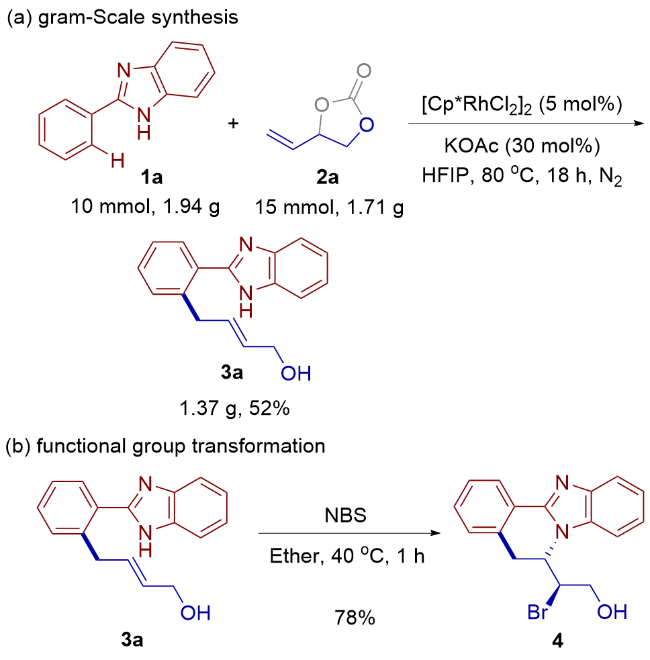
2.4 反应机理
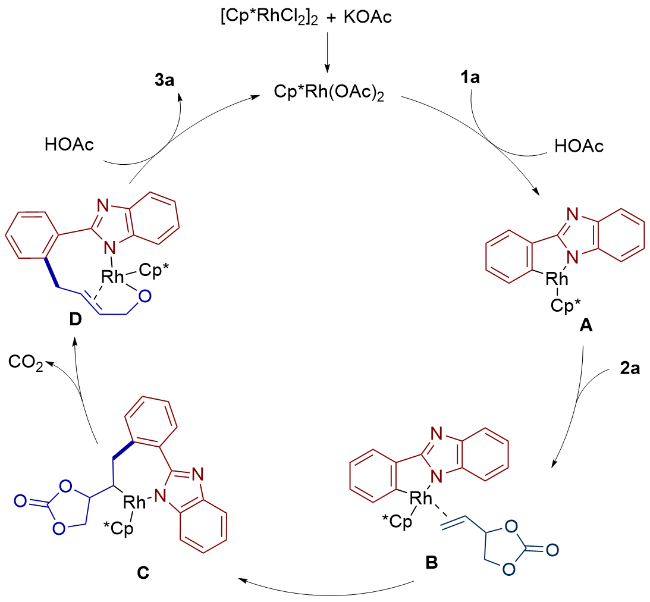
本文提出了可能的反应机理如图4所示. 首先, 催化剂[Cp*RhCl2]2与KOAc通过配体交换生成活性物种Cp*Rh(OAc)2. 随后, 该活性催化剂与2-芳基苯并咪唑(1a)发生协同金属化-去质子化(CMD)过程, 形成关键的铑杂环中间体A. 中间体A与4-乙烯基-1,3-二氧戊环-2-酮(2a)配位产生中间体B, 经迁移插入反应生成七元环铑中间体C[13d]. 根据已有的文献报道, 中间体C可能通过反式β-氧消除释放一分子CO2, 形成含有与Rh中心配位的反式C=C键的十元环铑中间体D. 但基于我们的实验结果, 不能排除顺式β-氧消除生成顺式产物的可能性. 最终, 中间体D在HOAc作用下质子化得到目标产物3a, 同时再生Cp*Rh(OAc)2催化剂完成催化循环.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
本文成功开发了一种铑(III)催化的2-芳基苯并咪唑与易得原料4-乙烯基-1,3-二氧戊环-2-酮的C—H烯丙基化反应的新方法. 该策略具有以下显著优势: 反应条件温和, 可规模化放大反应, 且对各类官能团均表现出良好的兼容性, 无需保护基即可实现结构多样性底物的高效偶联. 基于2-芳基苯并咪唑骨架在药物化学中的重要性, 这一高效转化策略有望为具有生物活性和合成价值的烷基化2-芳基苯并咪唑衍生物的制备提供新途径.
4 实验部分
向反应管中依次加入2-芳基苯并咪唑(1a~1r) (0.2 mmol, 1.0 equiv.)、[RhCp*Cl2]2 (6.2 mg, 0.01 mmol, 5 mol%)、KOAc (5.9 mg, 0.06 mmol, 30 mol%)、4-乙烯基-1,3-二氧戊环-2-酮(2a) (34.2 mg, 0.3 mmol, 1.5 equiv.)以及溶剂HFIP (1.5 mL). 反应体系在氮气保护下于80 ℃搅拌反应18 h. 反应完成后, 用乙酸乙酯(EtOAc) (5 mL)稀释反应液, 减压浓缩得到粗产物. 粗产物经快速柱层析分离[V(正己烷)∶V(EtOAc)=2∶1], 得到相应的白色固体产物3a~3r, 其收率为25%~61%.
采用2-芳基苯并咪唑(1a)进行克级规模反应: 向反应管中依次加入2-芳基苯并咪唑(1a) (1.94 g, 10 mmol, 1.0 equiv.)、[RhCp*Cl2]2 (124 mg, 0.2 mmol, 2 mol%)、KOAc (295 mg, 3 mmol, 30 mol%)、4-乙烯基-1,3-二氧戊环-2-酮(2a) (1.71 g, 15 mmol, 1.5 equiv.)以及溶剂HFIP (15 mL). 反应条件和分离纯化操作与上述步骤相同, 得到白色固体产物3a (1.37 g), 收率52%.
将化合物3a (264.1 mg, 1.0 mmol, 1.0 equiv.), N-溴代丁二酰亚胺(NBS) (622.9 mg, 3.5 mmol, 3.5 equiv.)和乙醚(10 mL)加入到50 mL的厚壁耐压管中. 在40 ℃的油浴中反应1 h冷却至室温后, 减压除去溶剂. 生成的粗产物用硅胶柱纯化[V(二氯甲烷)∶V(甲醇)=30∶1], 得到相应的白色固体产物4, 其收率为78%.
Cheng, F.