

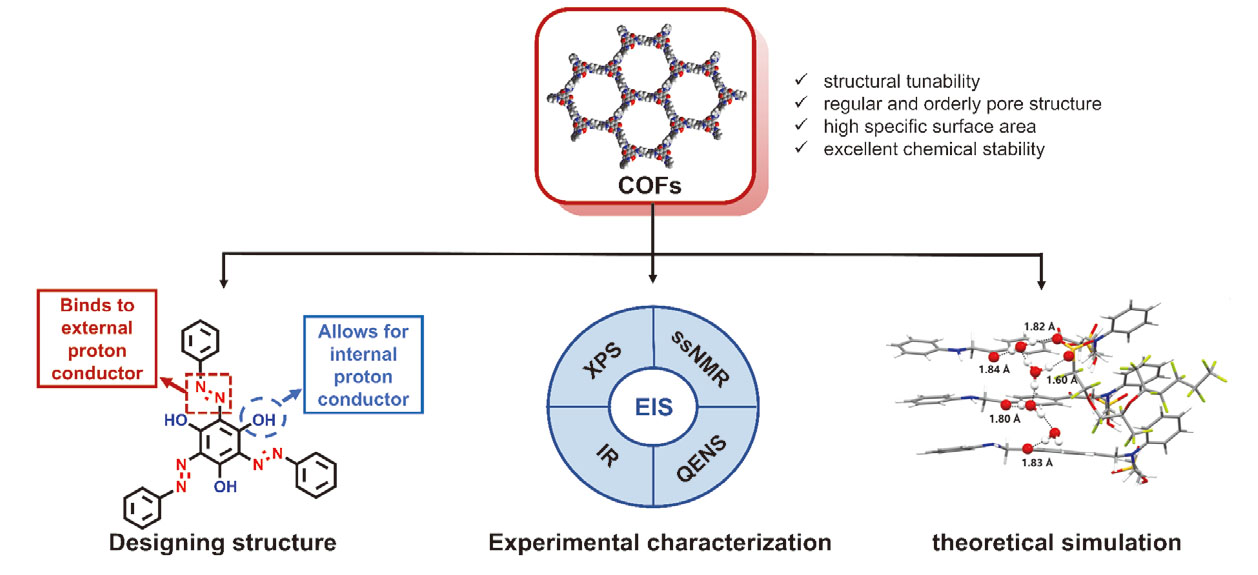
1 引言
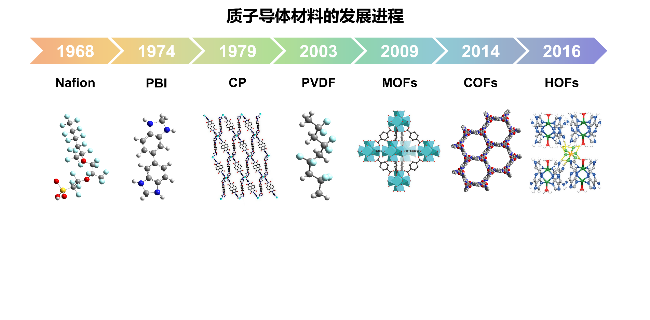
2 COF作为质子导体的研究进展
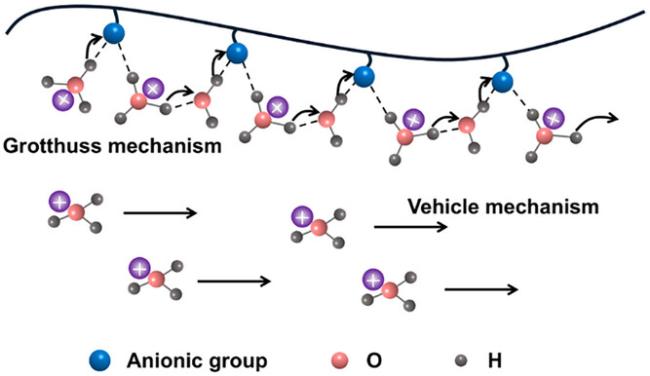
2.1 COF的质子传输机制
2.2 本征质子源
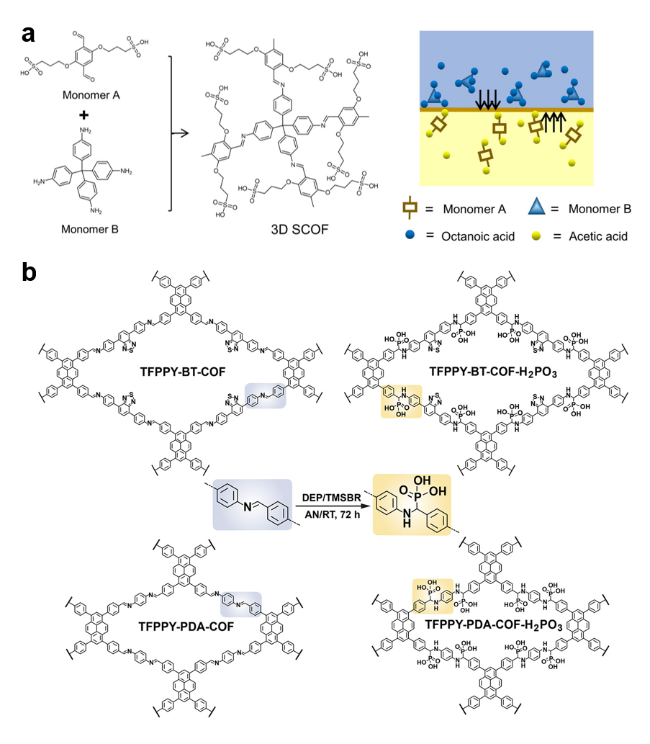
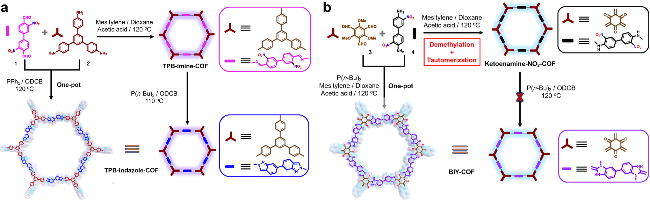
2.3 非本征质子源
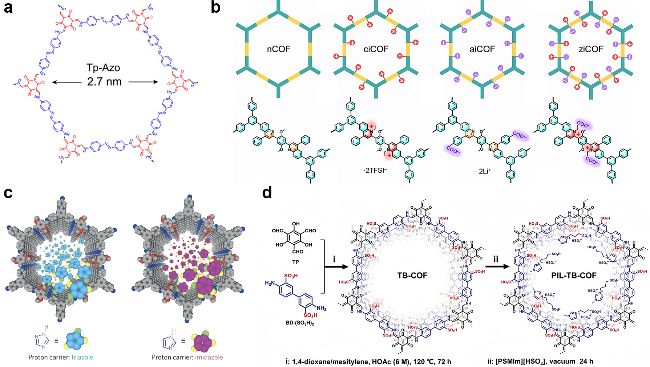
3 COF中质子传输的表征方法
3.1 质子传输速率的表征
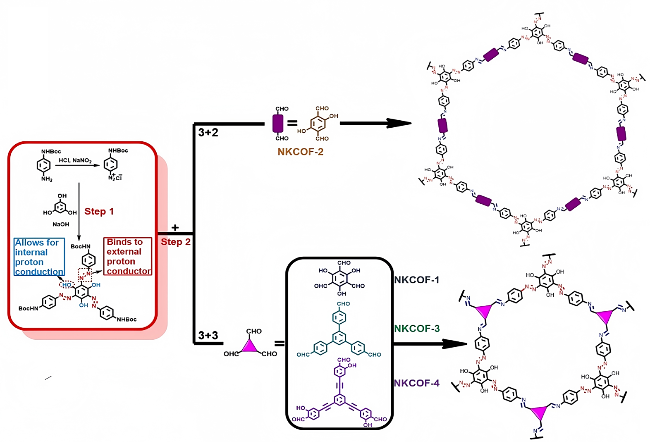
3.2 质子传输机制的表征
3.2.1 COF质子传输机制的表征方法
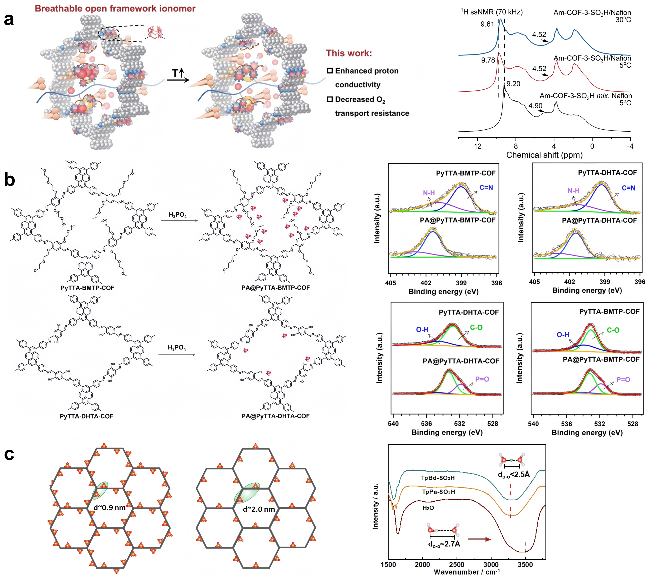
3.2.2 其他材料质子传输机制的表征方法
4 COF质子传输的模拟方法
4.1 密度泛函理论
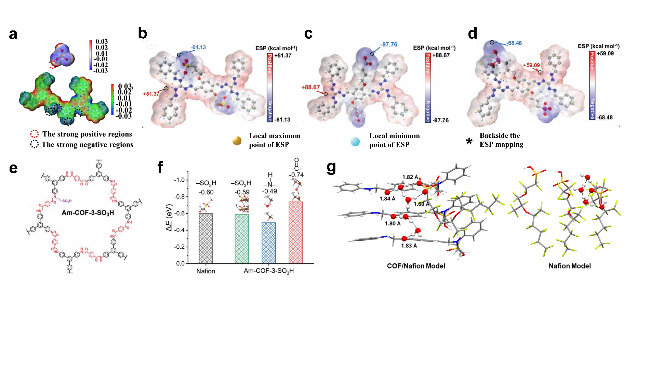
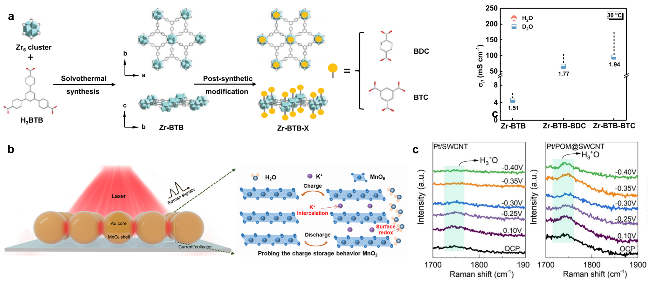
图9 (a) H3PO4和NKCOF-54的静电势等值面[70]. (b) ziCOF-1, (c) ziCOF-2和(d) ziCOF-3的静电势图[71]. (e) AM-COF-3-SO3H单元结构示意图. (f) 水分子在Nafion(-SO3H)和Am-COF-3-SO3H(-SO3H, N—H和C=O)不同位点的结合能. (g)四个水分子分别在COF/Nafion复合模型(左)和Nafion模型(右)的吸附情况[61]Figure 9 (a) Electrostatic potential isosurface of H3PO4 molecule and NKCOF-54[70]. The electrostatic potential maps of (b) ziCOF-1, (c) ziCOF-2 and (d) ziCOF-3[71]. (e) Illustration of AM-COF-3-SO3H unit structure. (f) Binding energy of a water molecule at different sites within Nafion (-SO3H) and Am-COF-3-SO3H (-SO3H, N—H, and C=O). (g) Adsorption of four water molecules on the COF/Nafion composite model (left) and Nafion model (right)[61] |

图10 孔隙内各向异性H+迁移行为的(a)理论阐明以及(b)质子迁移能量图[72] (IS、IMS和FS分别代表初始状态、中间状态和最终状态)Figure 10 (a) Theoretical elucidation and (b) proton migration energy diagram of anisotropic H-ion migration behaviors within the pore[72] (IS, IMS, and FS stand for the initial state, intermediate state, and final state, respectively) |
4.2 经典分子动力学模拟方法
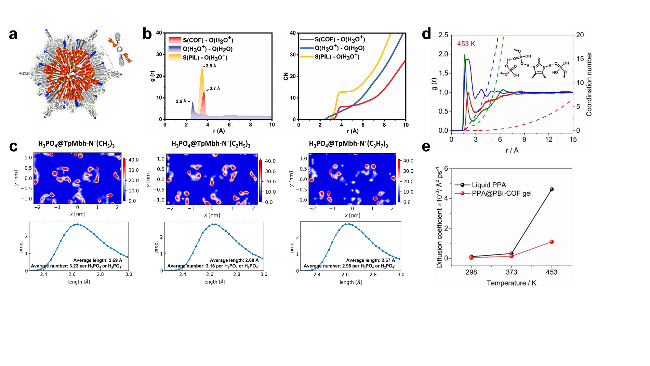
图11 (a)锁有H3PO4网络的1D通道H3PO4@TPB-DMeTP-COF重建晶体结构(俯视图)[73]. (b) S(COF)-O(H3O+)、O(H2O)-O(H3O+)和S(PIL)-O(H3O+)的RDF和CN[74]. (c) H3PO4@QACOFs的H3PO4密度分布(上), 以及H3PO4@QACOFs中形成的氢键的平均长度和氢键的平均数量大小(下)[75]. (d) PPA@PBI-COF中氢键的RDF和配位数. (e) PPA和PPA@PBI-COF的温度依赖性扩散系数[76]Figure 11 (a) Reconstructed crystal structure of H3PO4@TPB-DMeTP-COF (top view) with the H3PO4 network locked in the 1D channel[73]. (b) RDF and CN of S(COF)-O(H3O+), O(H2O)-O(H3O+), and S(PIL)-O(H3O+)[74]. (c) The obtained H3PO4 density profiles for H3PO4@QACOFs (top), and the average length of H-bonds and average number of H-bonds formed by H3PO4@QACOFs (down)[75]. (d) RDF and coordination number of the hydrogen bond in PPA@PBI-COF. (e) Temperature-dependent diffusion coefficients of liquid PPA and the PPA@PBI-COF[76] |
4.3 从头算分子动力学模拟方法
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
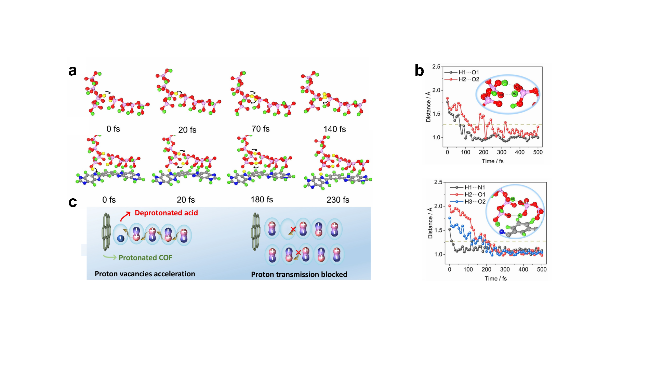
图12 (a) AIMD模拟中PPA和PPA@PBI-COF快照结构. (b) AIMD模拟中PPA和PPA@PBI-COF选定原子之间的时间依赖距离. (c)质子空位加速质子传导的机理示意图[76]Figure 12 (a) Snapshot structures taken from the AIMD simulation of PPA and PPA@PBI-COF. (b) Time-dependent distances between selected atoms in PPA and PPA@PBI-COF from the AIMD simulation. (c) Schematic diagram of the mechanism of proton vacancies accelerating proton conduction[76] |