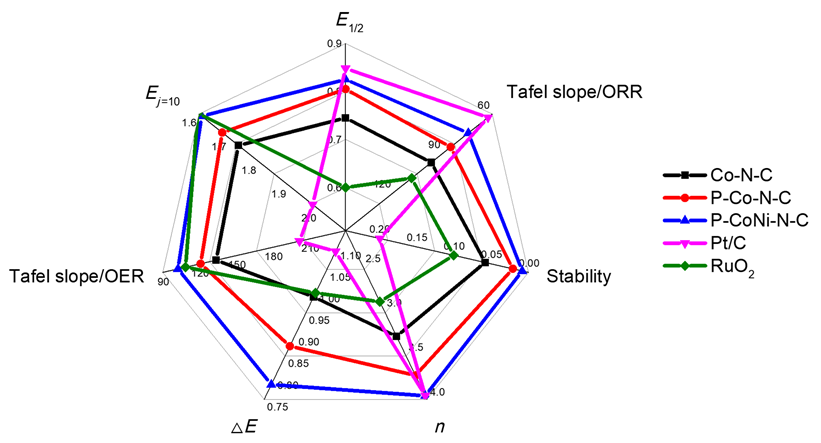
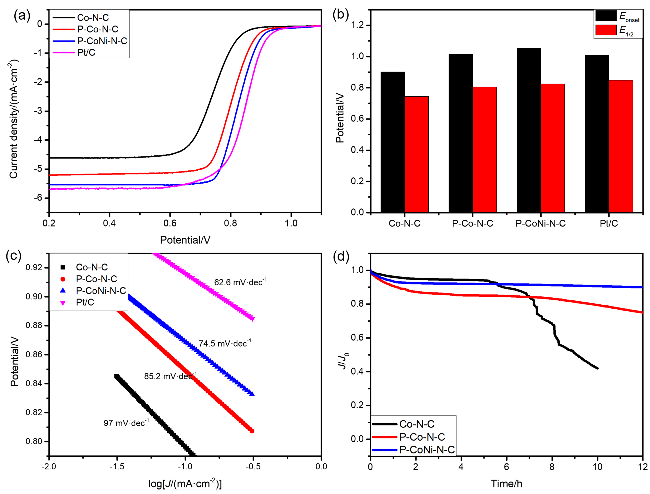
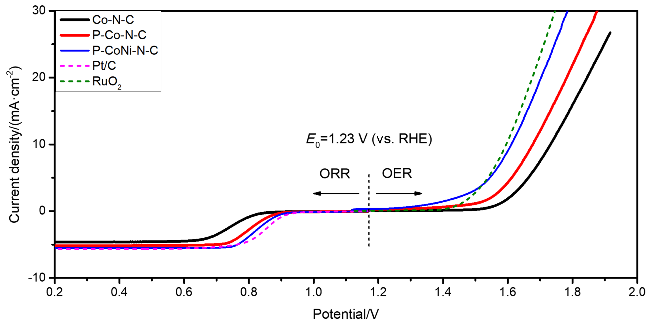
接着, 系统考察了Co-N-C、P-Co-N-C、P-CoNi-N-C及商用RuO
2四种催化剂的OER性能.
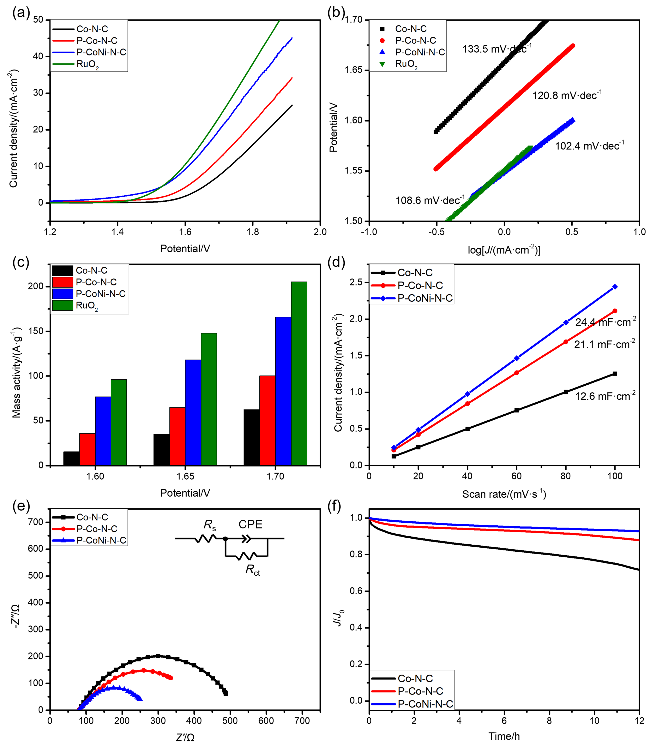
图8(a)显示了Co-N-C、P-Co-N-C、P-CoNi-N-C及商用RuO
2催化剂的OER极化曲线, 其中将电流密度为10 mA•cm
-2时的电位(
Ej=10)和该电位下的过电势(
η)作为衡量催化剂OER活性的重要指标. P-CoNi-N-C的
Ej=10显示为1.609 V (vs. RHE), 对应过电位为379 mV, 明显低于Co-N-C (1.732 V,
η=502 mV)和P-Co-N-C (1.678 V,
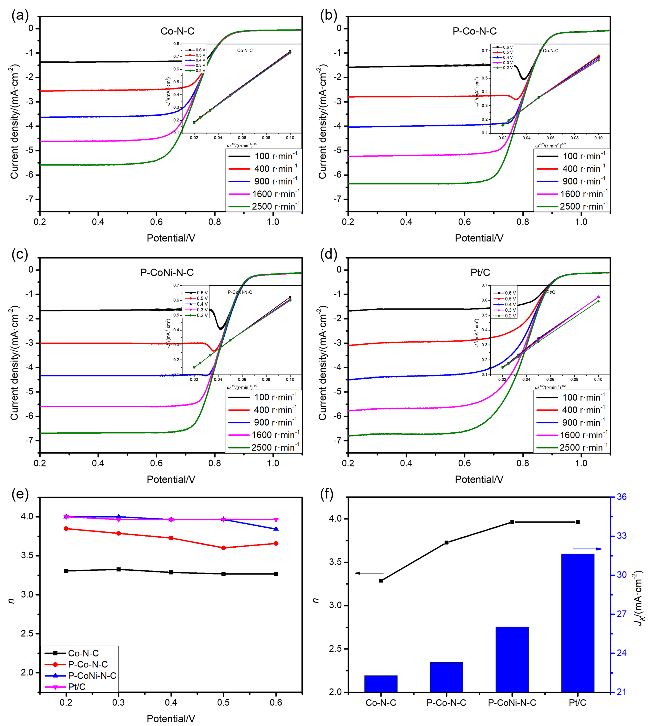
η=448 mV). 之后, 对催化剂的稳态动力学速率进行研究, 如
图8(b)所示, Co-N-C、P-Co-N-C、P-CoNi-N-C及商用RuO
2催化剂的Tafel斜率值分别为133.5、120.8、102.4和108.6 mV•dec
-1, 其中P-CoNi-N-C具有最低的Tafel斜率, 也被证明产生了更快的OER反应动力学过程. 质量活性(MA)测试表明(
图8(c))在1.60 V电势下P-CoNi-N-C的MA为76.95 A•g
-1, 达到RuO
2的80% (96.09 A•g
-1), 较Co-N-C (15.61 A•g
-1)和P-Co-N-C (35.82 A•g
-1)提升2~5倍. 此外, 为了评估参与电化学催化反应的实际表面积, 采用循环伏安法分别以10、20、40、60、80和100 mV•s
-1的扫描速率进行测试, 根据电化学活性表面积(ECSA)确定Co-N-C、P-Co-N-C和P-CoNi-N-C三种催化剂的双电层电容值(
Cdl). 如
图8(d)所示, Co-N-C、P-Co-N-C和P-CoNi-N-C的
Cdl值分别是12.6、21.1和24.4 mF•cm
-2. 显然, P-CoNi-N-C具有最高的
Cdl值, 表明P-CoNi-N-C的分级多孔结构、双金属掺杂以及独特的异质界面有利于其暴露更多的OER活性位点, 并促进了其析氧行为. 电化学阻抗谱图及其等效电路如
图8(e)所示, 与Co-N-C和P-Co-N-C两者相比, 显然P-CoNi-N-C具有更小的半圆直径, 证实了其具有较低的电荷转移电阻和优化的电子传输网络. 同样地, 采用计时电流法评价了Co-N-C、P-Co-N-C、P-CoNi-N-C的OER稳定性. 如
图8(f)所示, P-CoNi-N-C的电流保留率在12 h后仍可达到初始电流的92%, 而Co
3O
4在12 h后已经下降到71%左右, 以上表明P-CoNi-N-C具有长期高活性运行的潜力.


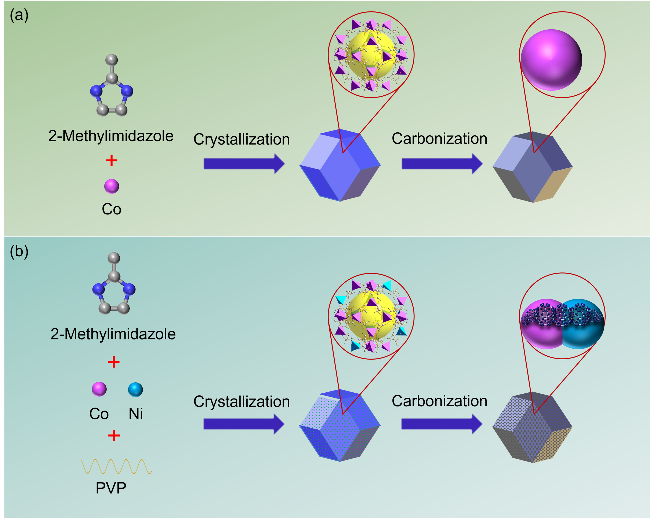


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}