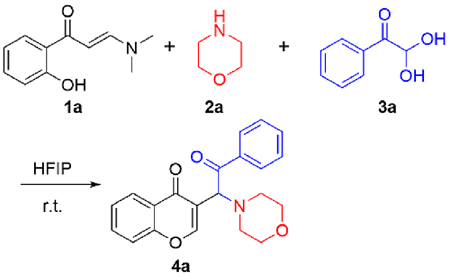
在不添加任何催化剂的条件下, 分别使用不同的溶剂, 如二氯甲烷(DCM), 甲苯(Toluene), 异丙醇(IPA), 1,4-二氧六环(1,4-Dioxane), HFIP,
N,
N-二甲基甲酰胺(DMF), 常温反应12 h, 则以HFIP为溶剂时, 得到相应的产物
4a产率较高, 为55% (Entries 1~6). 若HFIP与其他溶剂(如甲苯)组成的混合溶剂进行反应, 不如单独使用HFIP的效果好(Entry 5 vs. Entry 7). 同时, 研究还发现, 若提高吗啉
2a的投料比例至3 equiv.时,
4a产率显著提高, 可达70% (Entry 9). 鉴于HFIP当溶剂时, 有作为布朗斯特酸促进反应的效果, 我们尝试在反应体系中分别加入20 mmol%的路易斯酸Sc(OTf)
3或Y(OTf)
3、质子酸三氟乙酸(TFA)、三氟甲磺酸(TfOH),
4a的产率没有提高(Entry 9 vs. Entries 10~13). 研究发现, 调整投料顺序, 先加入
2a和
3a搅拌反应0.5 h, 再加入
1a反应12 h,
4a的产率可提高至75% (Entry 14). 若
2a和
3a一起加入室温反应0.5 h后, 再将烯胺酮
1a均分五次添加(每次添加量相等), 相邻两次添加间隔20 min, 则
4a的收率可提高至85% (Entry 15). 提高温度并不能提高反应收率(Entry 16). 因此, 本研究选用
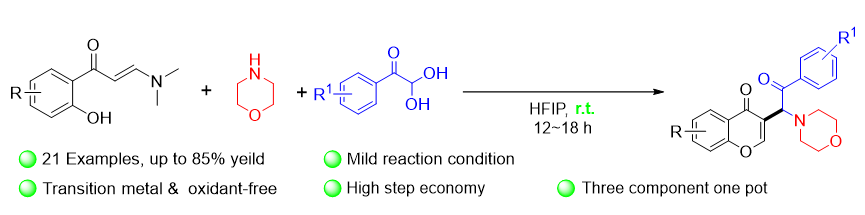
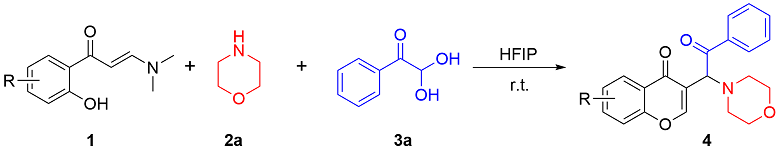
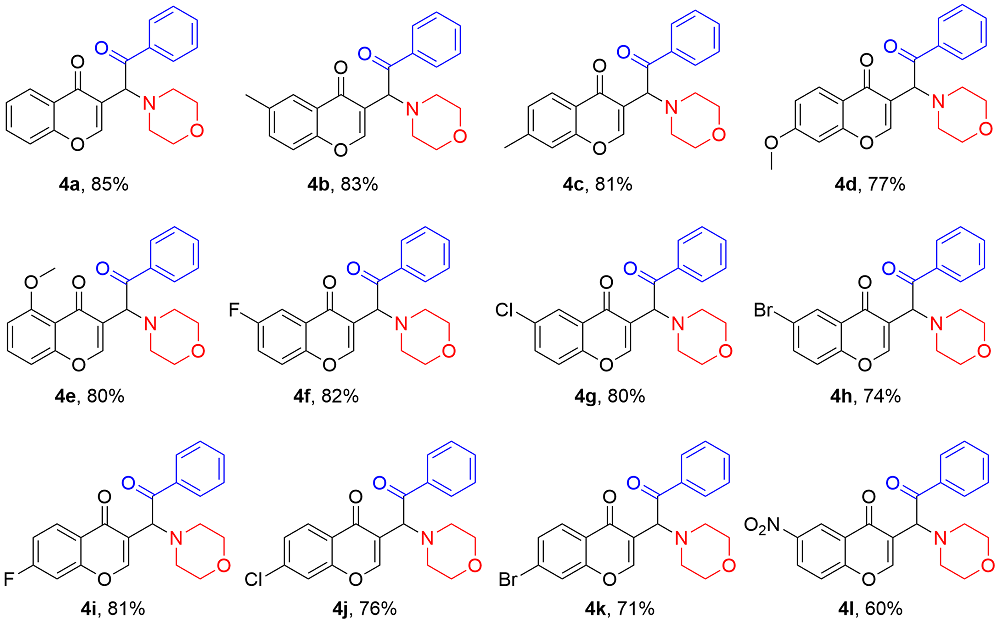
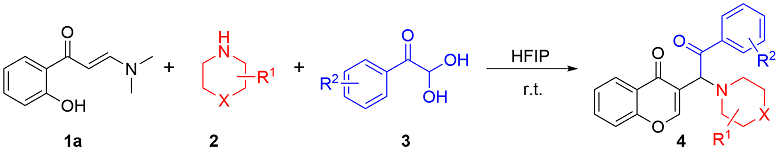
表1, Entry 15中的条件作为该反应的最优条件.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}