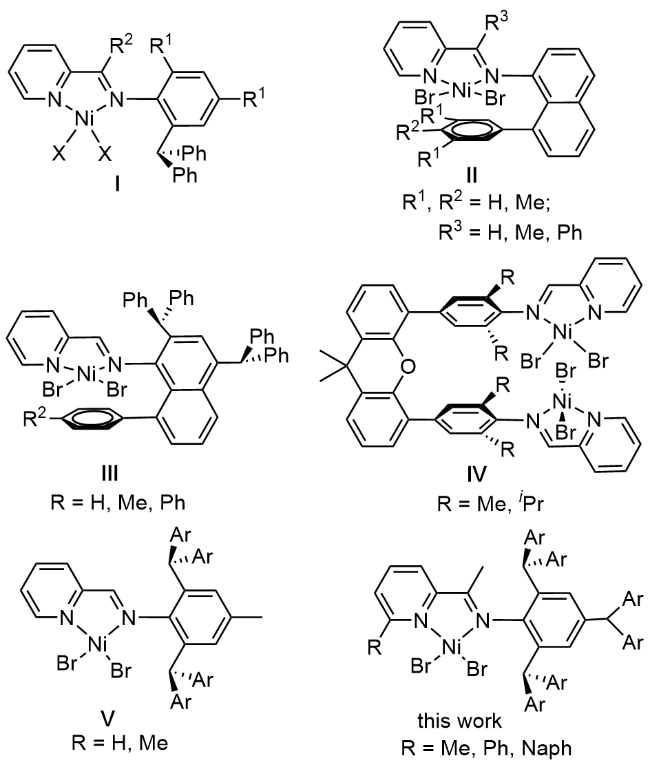
配体L1的合成: 称取2,4,6-三(双(4-氟苯基)甲基)苯胺(7.00 g, 10 mmol), 乙酰基2-甲基吡啶(1.49 g, 11 mmol), 少量对甲苯磺酸于250 mL圆底烧瓶中, 加入100 mL甲苯后置于磁力搅拌器上, 在120 ℃下充分搅拌12 h. 反应结束后, 冷却至室温并真空浓缩多余甲苯, 用甲醇重结晶并洗涤数次, 真空下干燥得到青色固体即配体L1 (4.98 g, 收率为61%). 1H NMR (400 MHz, DMSO-d6) δ: 7.73 (d, J=2.0 Hz, 1H, Ar-H), 7.70 (d, J=7.8 Hz, 1H, Ar-H), 7.33 (dd, J=6.9, 1.8 Hz, 1H, Ar-H), 7.09~6.80 (m, 24H), 6.42 (s, 2H, Ar-H), 5.50 (s, 1H), 5.19 (s, 2H), 2.48 (s, 3H), 1.26 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 161.87, 161.84, 161.82, 159.45, 159.43, 159.40, 156.77, 154.48, 145.90, 139.96, 139.93, 138.86, 138.83, 138.17, 138.14, 137.28, 130.49, 130.42, 130.35, 128.47, 124.62, 118.06, 115.19, 115.00, 114.98, 114.89, 114.79, 114.68, 53.09, 49.84, 24.05, 17.02. Anal. calcd for C53H38F6N2: C 77.93, H 4.69, N 3.43; found C 77.95, H 4.68, N 3.39. HRMS calcd for C53H38F6N2 [M+H]+817.3020, found 817.3009.
配体L2的合成: 采用与L1相同的合成方法, 得到青色固体即配体L2 (5.58 g, 收率为63%). 1H NMR (400 MHz, Chloroform-d) δ: 8.05 (d, J=7.6 Hz, 2H, Ar-H), 7.93 (d, J=7.1 Hz, 1H, Ar-H), 7.84 (q, J=7.3 Hz, 2H, Ar-H), 7.56~7.42 (m, 3H, Ar-H), 6.88 (dt, J=12.8, 7.0 Hz, 24H), 6.48 (s, 2H, Ar-H), 5.31 (s, 1H), 5.24 (s, 2H), 1.29 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 162.73, 160.28, 155.98, 139.76, 138.89, 138.78, 137.97, 137.28, 132.64, 130.69, 130.61, 130.54, 129.43, 129.18, 128.97, 126.86, 121.41, 119.58, 115.54, 115.33, 115.24, 115.08, 115.03, 114.87, 54.64, 50.65, 17.57. Anal. calcd for C58H40F6N2: C 79.26, H 4.59, N 3.19; found C 79.22, H 4.53, N 3.17. HRMS calcd for C58H40F6N2 [M+H]+879.3180, found 879.3172.
配体L3的合成: 采用与L1相同的合成方法, 得到青色固体即配体L3 (8.80 g, 收率为94%). 1H NMR (400 MHz, Chloroform-d) δ: 8.45~8.35 (m, 2H, Ar-H), 8.23~8.13 (m, 3H, Ar-H), 7.97~7.78 (m, 5H, Ar-H), 7.19~7.10 (m, 24H), 6.76 (s, 2H, Ar-H), 5.58 (s, 1H), 5.53 (s, 2H), 1.56 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 168.91, 159.15, 153.97, 145.50, 138.65, 138.62, 137.86, 137.83, 136.94, 136.64, 135.80, 133.04, 131.26, 129.88, 129.80, 129.55, 129.47, 129.45, 129.37, 126.68, 125.37, 124.97, 124.58, 124.27, 118.12, 114.32, 114.11, 114.07, 113.92, 113.86, 113.71, 53.51, 49.51, 16.44. Anal. calcd for C62H42F6N2: C 80.16, H 4.56, N 3.02; found C 80.14, H 4.54, N 3.01. HRMS calcd for C62H42F6N2 [M+H]+929.3330, found 929.3316.
镍催化剂Ni1~Ni3由相应的配体与NiBr2(DME)在CH2Cl2中络合反应合成. Ni1~Ni3的典型合成方法如下: 在手套箱中, 称取NiBr2(DME) (0.31 g, 1.00 mmol)、配体(1.00 mmol)和无水CH2Cl2 (20 mL)于史莱克瓶中, 在氮气气氛下室温反应16 h. 反应结束后, 过滤悬浮液并真空浓缩除去溶剂, 加入正己烷重结晶, 抽滤后使用正己烷洗涤固体三次, 真空干燥得到固体粉末.
Ni1为棕色固体(0.46 g, 73%), 元素分析C53H38Br2F6N2Ni理论值: C 61.48, H 3.70, N 2.71; 实测值: C 61.47, H 3.72, N 2.73. HRMS calcd for C53H38F6N2NiBr [M+H-Br]+ 954.148, found 954.988.
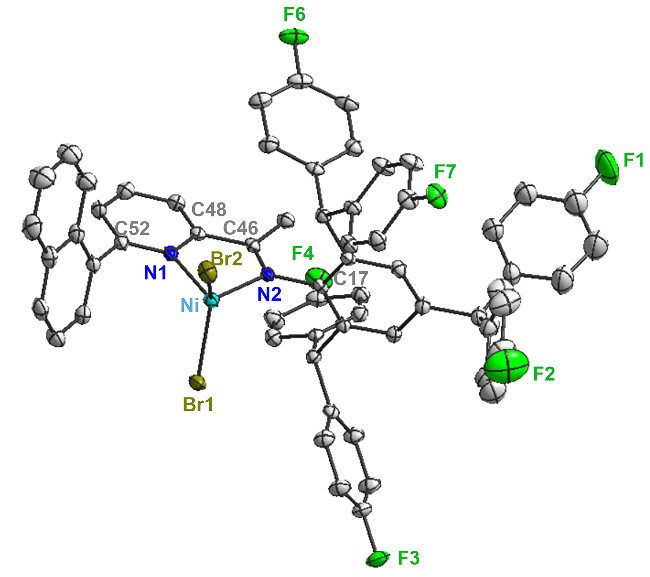
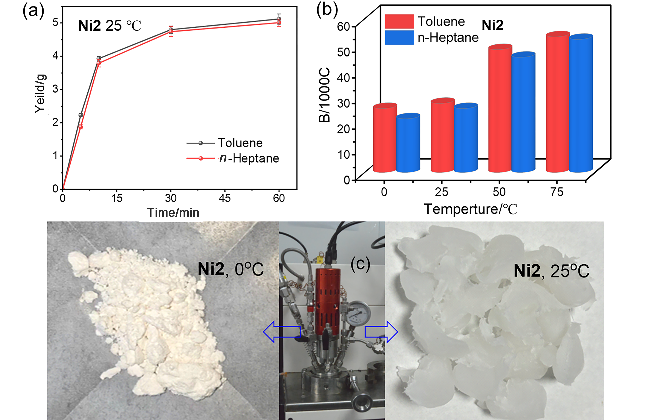
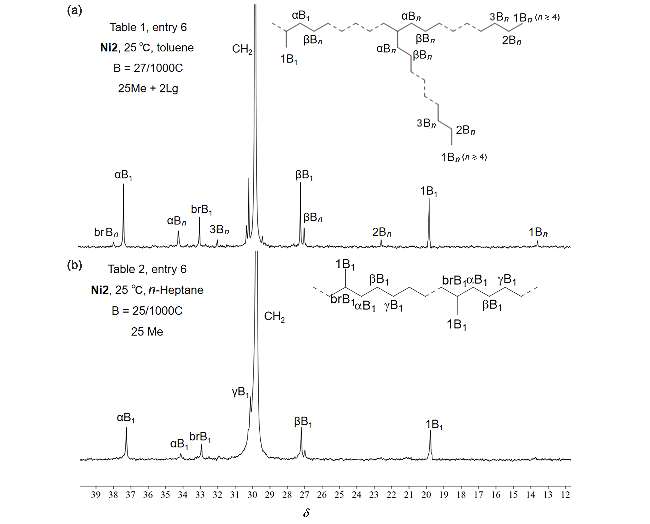
Ni2为棕色粉末(0.53 g, 79%), 元素分析C58H40Br2F6N2Ni理论值: C 63.48, H 3.67, N 2.55; 实测值: C 63.52, H 3.66, N 2.51. HRMS calcd for C58H40F6N2NiBr [M+H-Br]+ 1016.164, found 1016.9270.
Ni3为棕色固体(0.57 g, 86%), 元素分析C62H42Br2F6N2Ni理论值: C 64.89, H 3.69, N 2.44. 实测值: C 64.93, H 3.70, N 2.46. HRMS calcd for C62H42F6N2NiBr [M+H-Br]+ 1066.606, found 1066.9190.




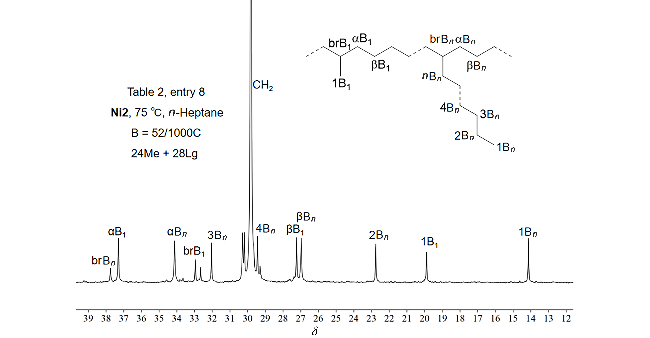
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}