

1 引言
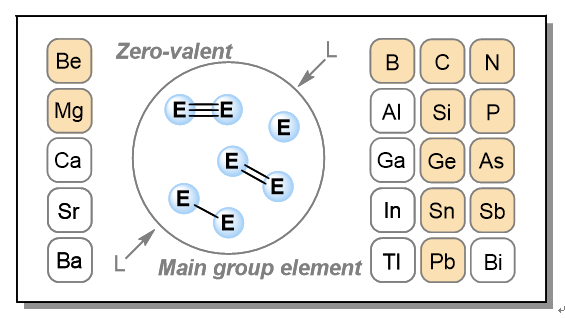
零价主族元素通常以单质形式存在. 由于原子排列方式的差异, 各元素单质可形成具有不同物理化学性质的同素异形体. 例如, 碳的同素异形体包括石墨、金刚石和富勒烯等, 氧的同素异形体有氧气和臭氧等, 磷的同素异形体则包含白磷、红磷和黑磷等. 不同键合方式赋予同素异形体独特的物理化学性质, 使其在光电材料等领域备受关注. 目前, 碳同素异形体的深入研究, 极大地加速了纳米技术和光电领域的发展[1]. 除此之外, 一些13至15族的原子簇化合物中, 也含有形式上的零价态元素, 它们具有独特的电子特性, 在材料科学领域具有重要的潜在应用价值[2]. 对于过渡金属, 利用配体的络合稳定作用, 许多单核的零价态过渡金属可以在室温下稳定存在, 例如Ni(CO)4、Cr(C6H6)2和Pd(PPh3)4等, 这些配合物作为催化剂在合成化学中具有重要应用. 然而, 配体稳定的单核或双核主族元素化合物却极为少见, 且难以在实验室条件下分离, 早期研究中仅能在极端条件下通过光谱学手段对其进行观测. 例如, 徐强和周鸣飞等[3-5]在低温基质隔离条件下, 对瞬态零价第14族元素羰基配合物En(CO)m (E=Si, Ge, Sn, Pb)及瞬态零价硼化合物B2(CO)2[6]进行了红外光谱表征. 此外, 周鸣飞和Frenking等[7-8]借助光谱学手段, 还观测到瞬态零价钙、锶、钡羰基配合物M(CO)8 (M=Ca, Sr, Ba), 以及瞬态零价铍氮气配合物Be(N2)n等物质. 因此, 稳定的单核及双核零价主族元素化合物的合成与分离是一项极具挑战性的研究课题, 在过去二十年中受到了日益广泛的关注.
卡宾(carbenes)是一类不符合八隅体规则的分子, 其中心二价碳原子既含一对孤对电子, 又具有一个空轨道, 目前已发展成为应用广泛的路易斯碱配体. 随着主族元素化学的发展, 卡宾已成为分离低价态主族元素的重要辅助配体[9], 借助不同类型的卡宾配体的稳定作用, 一些零价主族元素化合物的合成与分离得以实现, 并在过去的综述文章中被详细地总结[10-13]. 硅宾(silylenes)和锗宾(germylenes)配体作为卡宾的重元素类似物, 凭借其独特的电子结构、可灵活调节的空间位阻, 以及卓越的稳定性与反应性平衡特性, 不仅为分离更高活性物种提供了可能性, 还能通过与配位中心的协同作用参与化学反应, 开辟新的反应模式, 逐渐在低价主族元素化学领域展现出独特的优势(图式1). 本文从不同元素的单核及双核零价主族元素化合物的合成与分离出发, 涵盖了包括卡宾、硅宾和锗宾等在内的不同配体体系的使用, 侧重阐述这些零价主族元素化合物的结构特征与化学反应性质.
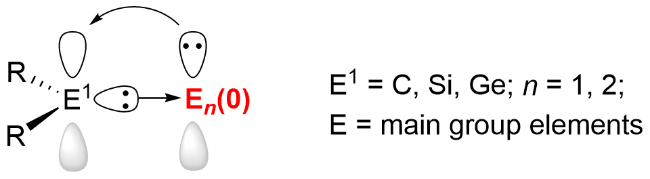
2 零价态碱土金属化合物的合成及反应性
2.1 零价铍化合物的合成及其反应性
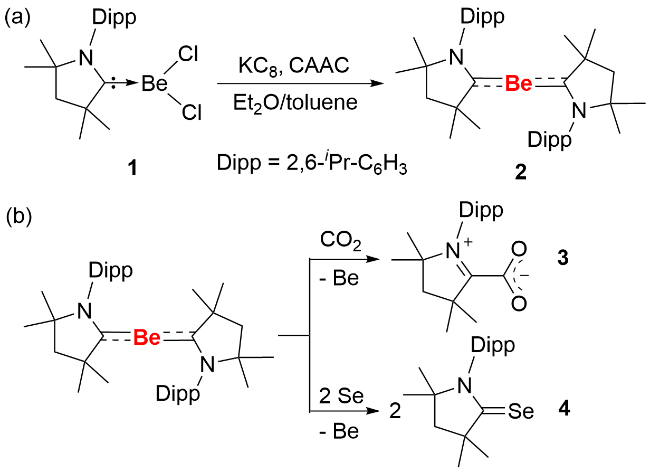
长期以来, 人们一直致力于低价态(零价和正一价)的铍化合物的合成研究, 作为碱土金属中最轻的元素, 铍具有较小的原子半径和相对较高的电负性, 这些特性使其能够形成较强的共价键. 因此, 理论上在适当条件下有望合成零价铍化合物. 此前, 零价铍化合物的研究多停留在理论预测阶段, 直到近年才取得了突破性进展. 2013年, Frenking等[14]通过理论计算预测了氮杂环卡宾(NHC)作为配体稳定单核零价铍的可行性. 2016年, Braunschweig等[15]利用环烷基氨基卡宾配体(CAAC)的稳定作用, 成功分离并表征了形式零价铍化合物(图式2a), 目前对该化合物结构认识仍存在争议[16-18]. 研究人员利用强还原剂石墨钾(KC8)与BeCl2的CAAC配合物1以及1 equiv.外加的CAAC配体进行反应, 分离得到紫色晶体2. 晶体结构显示, 化合物2结构中C—Be—C成直线型, C—Be键键长(0.165~0.166 nm)处于单键和双键键长范围之间, 明显小于其前体分子(0.177~0.179 nm). 理论计算表明, CAAC配体与中心铍原子间的电子供体-受体相互作用, 导致C—Be—C间存在三中心二电子π键, 且Be向CAAC配体的π反馈作用强于CAAC配体向Be的σ给电子作用, 这与CAAC配体的强π受体特性一致. 研究人员已对其反应活性开展初步探究, 结果表明, 化合物2与H2、硼烷及硼氢化物均不发生反应; 而与硒单质和CO2反应时, 会发生铍单质的脱落现象(图式2b).
2.2 零价镁化合物的合成及其反应性
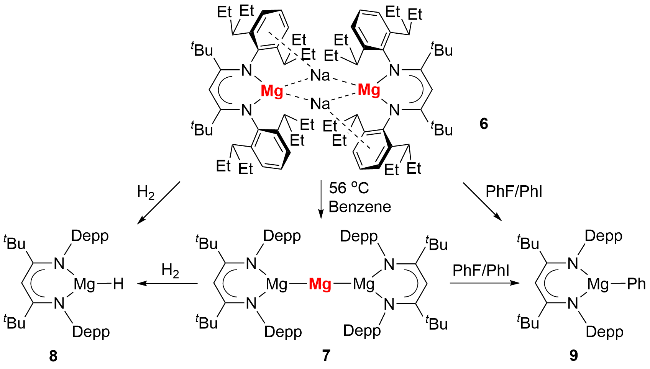
尽管有预测认为卡宾稳定的零价镁化合物在合成上不具备可行性[14], 研究人员仍探索通过其他途径实现零价镁化合物的分离. 2021年, Harder等[19]合成了首例具有极强还原能力的零价镁化合物(图式3)和 [Mg(I)-Mg(0)-Mg(I)]团簇化合物. 作者利用Na/NaCl还原二价镁前体(BDI*)MgI (BDI*=HC{C(tBu)N[2,6- (3-pentyl)-phenyl]}2) (5), 以48%的产率分离得到零价镁化合物6的红棕色晶体. 该化合物单晶结构显示其含有两个(BDI*)Mg结构基元, 并被两个Na离子桥连, Mg-Mg间距为0.578 nm, 表明二者间无明显成键作用. 通过对BDI*配体的空间结构及碳氢化学位移值的分析可推断, 不存在从镁到配体的电荷转移, 其中BDI*配体与镁的价态分别为-1价和0价. 自然布居分析(natural population analysis, NPA)进一步验证了上述结论, 故化合物6为形式上的零价镁化合物.
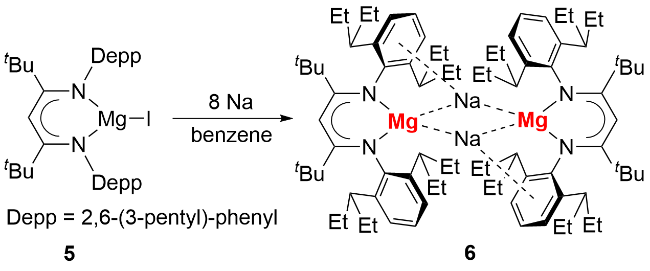
化合物6稳定性较差, 接触醚类溶剂或将钠离子替换为锂或钾离子均会导致分解. 令人意外的是, 6可以在56 ℃下加热转化, 以9%的产率分离得到深红色的(BDI*)Mg-Mg-Mg(BDI*)零价镁化合物7(图式4). 根据反应中生成的钠镜, 作者推测该化合物的生成经历极强还原性的零价镁对钠离子的还原过程. 反应化学探索发现, 6和7均可以和H2反应生成二价镁氢化合物8, 以及和氟苯或者碘苯反应生成(BDI*)MgPh (9).
3 零价态13族元素化合物的合成及反应性
3.1 双核零价硼化合物的合成及反应性
在早期研究中, 双核零价硼化合物(diborynes)仅能在极端条件下通过光谱学手段观测, 2002年, 周鸣飞等[6]在低温基质中通过光谱检测, 首次发现了羰基配体稳定的双核零价硼化合物(OCB≡BCO), 该化合物呈线型结构, 硼硼之间以叁键的形式结合在一起. 2012年, Braunschweig等[20]分离并表征了首例NHC稳定的双核零价硼化合物(图式5). 作者以钠/萘(Na(C10H8))为还原剂, 在四氢呋喃中还原四溴硼烷的NHC配合物10, 以57%的产率分离得到双核零价硼化合物11. 单晶结构显示, 化合物11中B—B—C键角约为173.0°, 接近直线型构型, B—B键长为0.1449 nm, 与理论计算预测的B≡B叁键键长接近. 11的核磁共振硼谱的化学位移为δ 39, 较还原前体10 (δ -4.8)相比向低场移动.

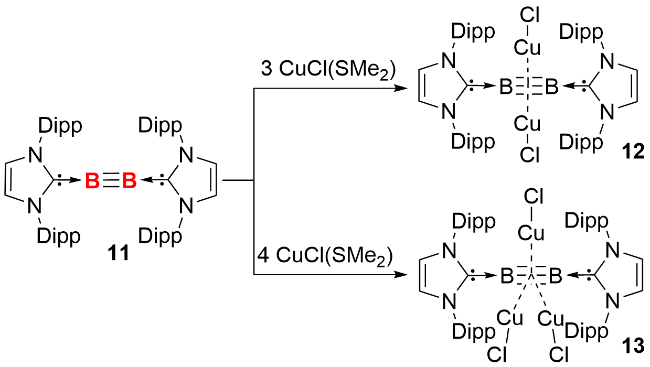
2014年, 该团队用CAAC配体替换NHC配体, 采用类似的合成途径, 分离得到CAAC配体稳定的双硼化合物15(图式7)[22]. 单晶结构显示, 化合物15同样呈现直线型构型, 但其B—B键(0.1489 nm)较化合物11有所延长, 而B—C键则变短(分别为0.1458 nm和0.1459 nm), 这反映了CAAC配体与NHC配体在电子性质上的显著差异. CAAC配体接受中心原子反馈p电子的能力更强, 导致上述键长的变化. 分子轨道(MOs)计算显示, NHC稳定的双核零价硼化合物的最高被占据轨道(HOMO)主要是B—B间的π键, 而CAAC配体稳定的双硼化合物的HOMO轨道则为C—B—B—C间的π体系, 与单晶结构键长变化一致. NPA计算表明, 化合物15的B2基团相对于NHC配位的B2更缺电子.
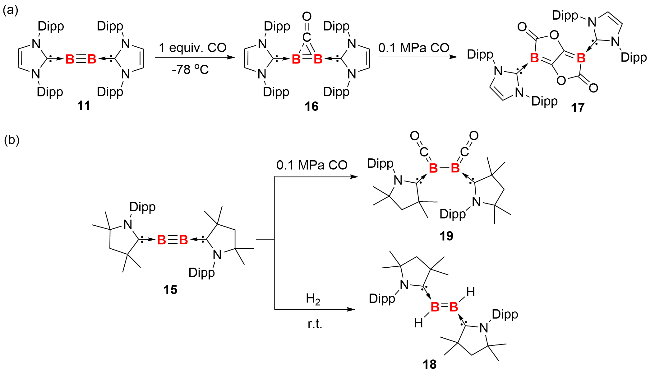
研究发现, 化合物11和15在与 H₂、CO 的反应中, 呈现出截然不同的化学行为. 在-78 ℃的低温环境下, 化合物11便能与CO发生反应, 首先生成CO桥联的双硼化合物中间体16[23]; 随后, 中间体16与过量CO进一步反应, 最终得到官能团化的双环双硼内酯化合物17(图式8). 相比之下, 化合物15与CO反应仅生成双硼烯酮化合物19, 且即便在高温高压条件下, 该化合物也不再与CO继续反应. 这表明随着卡宾和硼原子之间作用增强, 硼硼中心电子云密度降低, 致使双核零价硼化合物的还原性减弱, 对CO的反应活性也随之降低. 当这两种化合物与H2反应时, 表现出与CO反应相反的活性趋势: 化合物11在80 ℃下加热2 d, 都未与H2发生反应; 而化合物15则能在室温下与H2迅速反应, 生成二硼烯产物18.
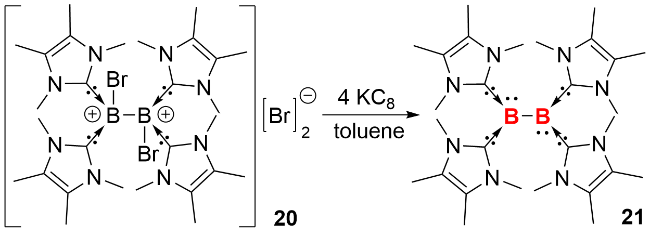
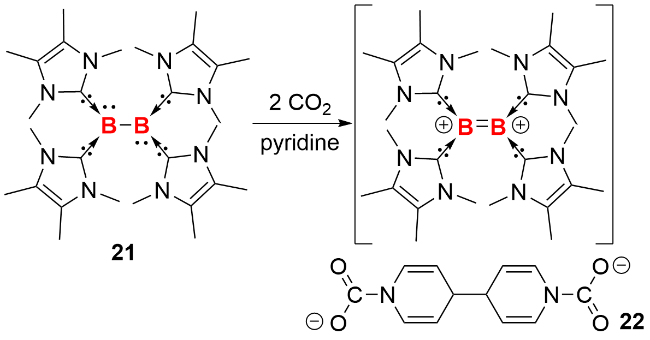
分子轨道计算结果表明, 化合物21的最高占据分子轨道(HOMO)和次高占据分子轨道(HOMO-1)分别对应B2基团上的1πg*和1πu轨道. 在反应化学研究中, 化合物21展现出独特的反应活性: 它能够与二氧化碳发生双单电子转移还原反应, 促使二氧化碳转化为两个二氧化碳自由基阴离子(${\text{CO}}_{2}^{·-}$), 同时B—B键转变为B=B双键. 生成的${\text{CO}}_{2}^{·-}$进一步将吡啶还原, 形成两个吡啶基-N-羧酸根自由基阴离子. 这些自由基阴离子在吡啶基自由基环的对位位置发生分子间C—C耦合, 最终生成4,4′-二氢联吡啶-二-N-羧酸盐二阴离子(图式10). 这一系列反应充分证明了化合物21的πg*反键轨道中两个电子的高活性, 展现出其独特的双单电子转移反应性.
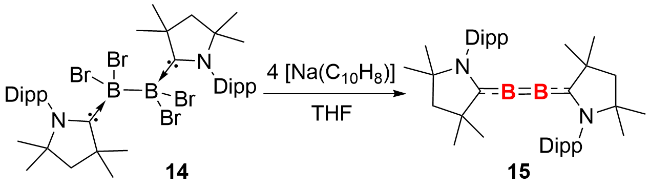
2024年, 我们利用脒基硅宾配体成功分离并表征了硅宾配体稳定的双核零价硼化合物24(图式11)[25]. 具体合成路径为: 在乙二醇二甲醚(DME)溶剂中, 使用KC8还原硅宾稳定的四溴化二硼23, 以68%的产率获得目标产物. 单晶结构分析显示, 化合物24的Si—B—B片段接近直线型(键角164.4°~177.4°), 其B—B键长(0.1444(4)~0.1459(3) nm)与卡宾稳定的双核零价硼化合物的B—B键长范围(0.1446~0.1465 nm)高度吻合. 分子轨道计算表明, 其HOMO与HOMO-1分别对应 B—B间两个正交的π成键轨道. 进一步通过自然键轨道理论(natural bond orbital theory, NBO)分析证实, B—B键由一个σ键(1.96 e)和两个π键(1.76 e、1.73 e)组成, Wiberg键级数为2.43, 明确揭示了该化合物的叁键特征.
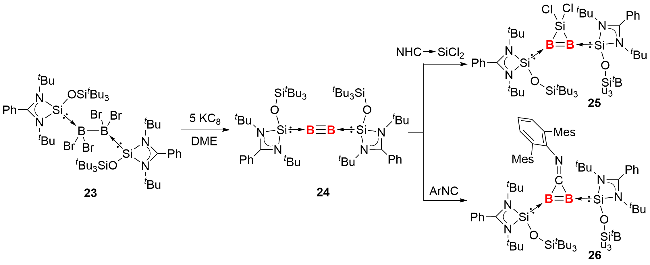
反应化学研究表明, 化合物24可与二价硅卤化物、异腈分别发生[1+2]环加成反应, 高效构筑结构新颖的杂原子环丙烯类似物——三元环硅杂二硼烯25和三元环二硼烯亚胺26. 单晶结构解析与理论计算显示, 两种三元环产物均具备一定的芳香性, 这一特性凸显了双核零价硼化合物在构建具有独特性能的硼杂环化合物领域的巨大应用潜力.
3.2 单核零价硼化合物的合成及其反应性
尽管双核零价硼的合成研究已有少数几例报道, 单核零价硼化合物的合成与分离却始终没有实现, 直至近期, Pranckevicius等[26]报道了CAAC 配体稳定的单核硼化合物的合成与表征(图式12). 作者首先在添加1 equiv. CAAC配体的条件下, 利用KC8还原三溴化硼的CAAC配合物27, 以38%的产率分离得到抗磁性的硼宾本体化合物(CAAC)2BH. 在对还原体系粗产物进行电子顺磁共振(EPR)表征时发现, 其中稳定存在一种顺磁性物质. 通过优化反应条件, 最终以72%的产率分离得到紫红色的单核硼化合物28. 单晶结构显示, 化合物28的C—B—C键角为176.03°, 呈近直线型结构, 其B—C键长(0.14464 nm和0.14471 nm)略长于典型的B—C双键, 表明硼原子与CAAC配体间存在强π相互作用.
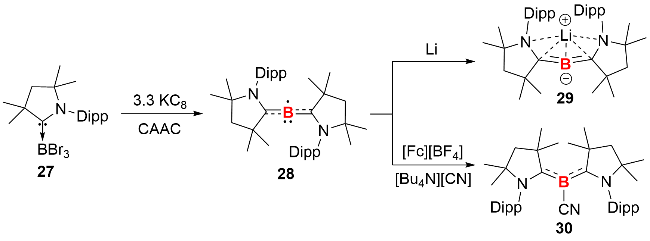
在甲苯中的EPR光谱显示, 未成对电子与11B和两个14N核的超精细耦合常数分别为0.45 G和4.34 G. 理论研究表明, 化合物28是自旋态为S=1/2的顺磁性物种, 自旋密度计算显示, 未成对电子在硼原子上的分布最多(占比为30%), 其余大部分自旋密度主要在两侧的两个卡宾碳原子(各20%)和两个氮原子(各10%)上.
作者进一步探究了化合物28的反应化学性质, 结果显示, 其在四氢呋喃中与锂单质发生还原反应时, 可以95%的高产率分离得到含两个B=C双键的 (CAAC)2BLi化合物29, 该产物可视为联烯的等电子体. 此外, 在氰根季铵盐([nBu4N][CN])存在下, 使用单电子氧化剂二茂铁(Ⅲ)四氟硼酸盐([Fc][BF4])对零价硼化合物28进行氧化, 可得到CAAC配体稳定的氰基硼宾化合物30.
4 零价态14族元素化合物的合成及反应性
4.1 零价碳化合物的合成及其反应性
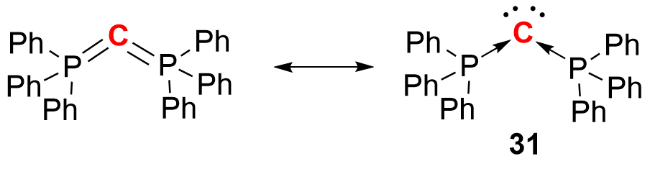
14族(碳族)元素的同素异形体在电子和光电材料领域具有重要地位. 配体稳定的零价14族元素化合物可视为新型同素异形体, 其分离与表征不仅有助于揭示零价态14族元素的成键方式, 而且其可溶性特点为合成化学带来了新的应用潜力. 然而, 由于零价14族元素本身具有较强的还原性, 且中心元素与配体间的结合作用有限, 这类化合物常面临聚合、歧化和分解等问题, 导致其合成与分离极具挑战性. 2006年, Frenking等[27]通过理论计算对碳二膦烷化合物C(PPh3)2的电子结构与成键方式展开研究. 该化合物由Ramirez等[28]于1961年首次合成, 最初被定义为双叶立德化合物. 然而, Frenking团队对C(PPh3)2的前线分子轨道分析显示, 其中心碳原子上存在两个孤对电子轨道: 一个是与HOMO对应的p型孤对轨道, 另一个是与HOMO-1对应的s型孤对轨道, 表明中心碳原子携带两对孤对电子. 基于此, C(PPh3)2被重新界定为单核零价碳化合物(carbones), 其结构中两个膦配体与中心碳原子通过电子供体-受体相互作用结合(图式13), 自此之后, 不同类型的零价碳化合物及其重元素类似物的合成与反应性研究相继被报道.
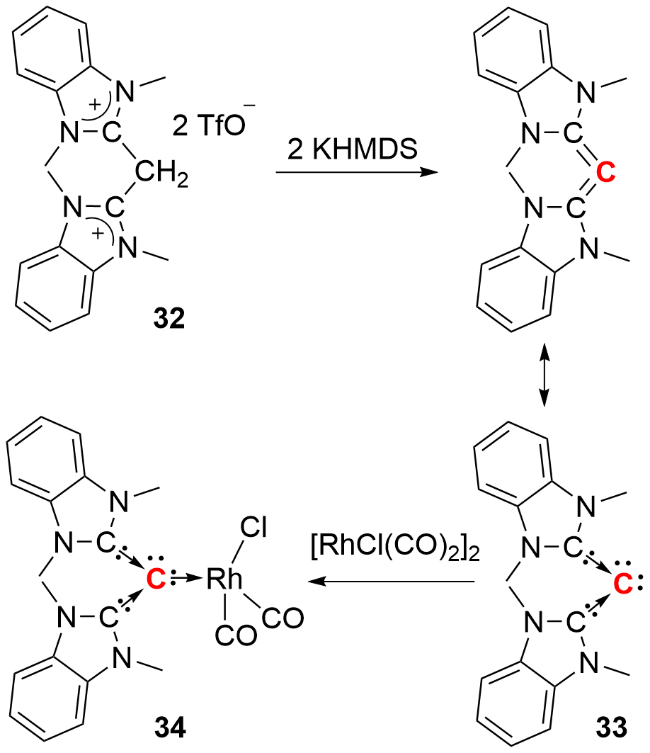
2007年, Bertrand等[29]报道了一种具有弯曲结构的开链型联烯化合物33 (carbodicarbene, C—C—C键角134.88°)的分离(图式14). 化合物33与[{RhCl(CO)2}2]反应时, 以56%的产率生成中心碳原子与Rh的σ配合物34; 值得注意的是, 常规烯烃与过渡金属配合物反应通常生成C=C双键的π配合物, 而该联烯化合物表现出独特的σ配位模式. 进一步分析显示, 配合物34的羰基伸缩频率平均值(2014 cm⁻1)显著低于NHC配体的相应配合物(2058~2036 cm⁻1), 表明该弯曲联烯化合物具有更强的给电子能力. 理论研究表明, 其中心碳原子上存在两对形式孤对电子, 赋予其比NHC配体更强的σ给电子能力和更弱的π接受能力, 因此化合物33可视为双卡宾稳定的单核零价碳化合物.
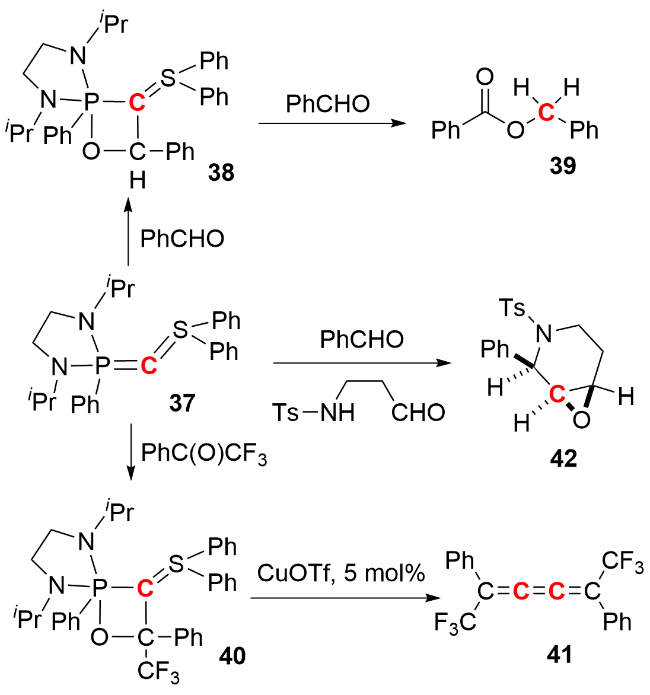
2010年, Kato和Baceiredo等[31]报道了不对称磷、硫-双叶立德化合物37 (mixed P,S-bis(ylide))的合成及其在多组分反应中作为零价单碳原子转移试剂的应用(图式16). 该化合物与1 equiv.苯甲醛反应生成含C2OP四元环的中间体38, 中间体38继续与另1 equiv.苯甲醛反应生成苯甲酸苄酯39并释放出化合物37; 当与1,1,1-三氟苯乙酮反应时, 生成类似的四元环状中间体40, 该中间体在催化量三氟甲磺酸亚铜(CuOTf)作用下进一步转化为累积烯烃41. 此外, 化合物37还可与苯甲醛及N-对甲苯磺酸-γ-氨基醛通过“一锅法”构建顺式环氧产物42. 在上述反应中, 化合物37的零价碳原子转移至有机底物, 分别形成sp3 仲碳、sp杂化碳及立体选择性季碳中心, 凸显了其作为零价单原子碳转移试剂在构建多元碳骨架中的潜在应用价值.
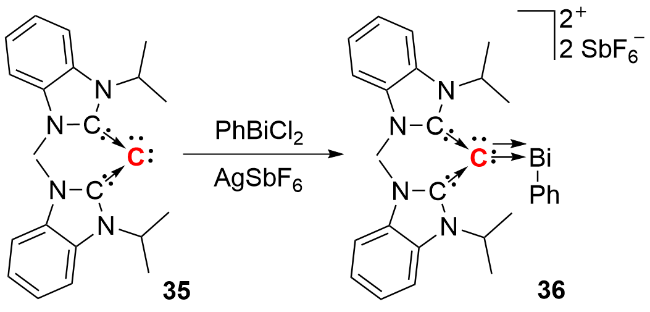
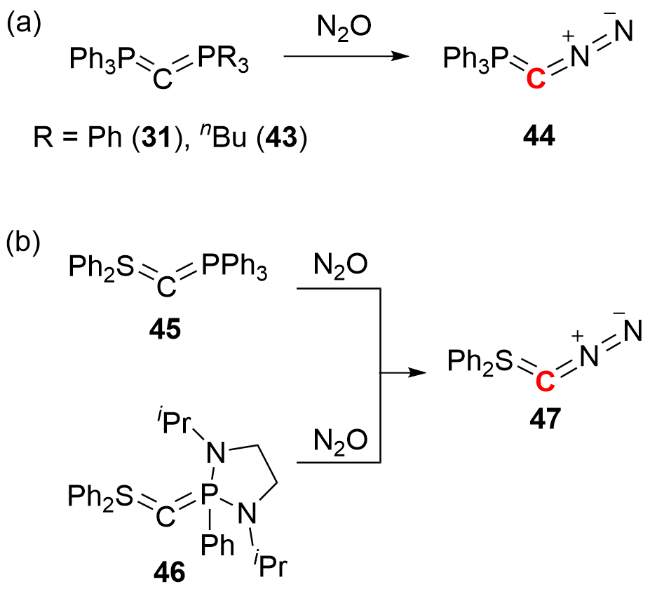
2024年, Hansmann等[32]报道了重氮磷叶立德化合物44 (diazophosphorus ylide)的合成与分离成果(图式17). 作者以碳二膦烷化合物31或43为前体, 使其与N2O反应, 通过重氮转移过程成功合成化合物44. 该化合物可以作为C(0)原子转移试剂, 用于构建多取代吡唑、炔烃以及累积烯烃等多种化合物. 紧接着在2025年, 该课题组通过类似的重氮转移途径, 报道了重氮硫叶立德化合物47 (diazosulfur ylide)的合成[33]. 该化合物可以与烯烃类化合物经过一步或分步反应, 实现四个 C—C键的构建, 构筑螺[2.2]戊烷和螺三环化合物等含sp3杂化碳原子的刚性螺环化合物, 这一系列成果丰富了零价碳化合物的应用范畴.
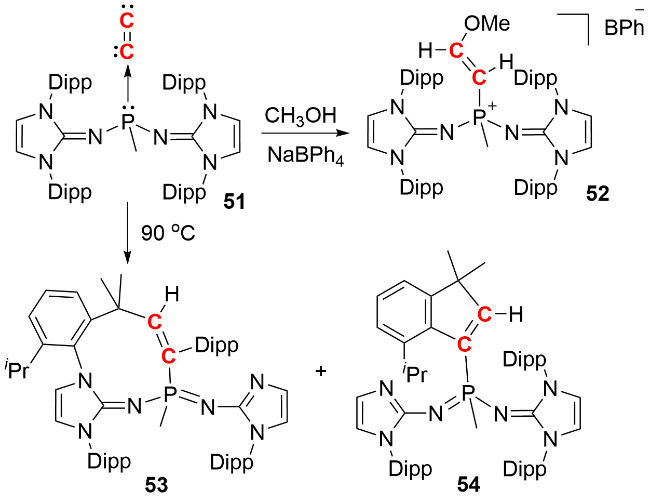
配体性质对双核碳化合物的电子结构与成键方式具有显著影响. 2021年, Ong和Frenking等[35]报道了利用大位阻双(咪唑啉亚胺基)甲基膦配体稳定的单配位双核碳化合物51的合成(图式19). 单晶结构分析表明, 其分子中含有非线型的P—C—C片段, C—C键长为0.1237 (4) nm, 介于传统C≡C叁键与未共轭双键之间. 化合物51在加热条件下可触发两种不同的活化反应: 一是异丙基的C—H键活化, 二是咪唑环取代基的C—N键活化, 分别生成环状化合物53和54. 当与甲醇反应时, 该化合物通过β-C对O—H键的插入作用生成产物52, 并伴随α-C的质子化. 上述反应特性表明, 化合物51的两个零价碳原子均表现出卡宾的反应活性.
4.2 零价硅化合物的合成及其反应性
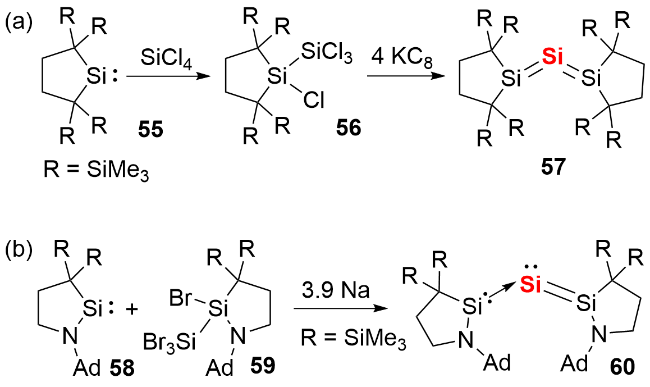
2003年, Kira等[36]报道了三硅烯化合物57的分离与表征(图式20a). 单晶结构显示, 其Si—Si—Si键角为 136.5°, Si—Si键长(0.2177 nm和0.2188 nm)处于Si=Si双键的键长范围, 作者最初将其视为联烯的硅类似物. 随后, Frenking等通过理论计算指出, 该化合物本质上是单核零价硅化合物(silylones), 硅原子间通过电子供体-受体相互作用成键. 2021年, Iwamoto等[37]利用环烷基胺基硅宾(CAASi)配体合成了类似的硅宾稳定零价硅化合物60(图式20b), 该化合物存在两种稳定异构体. 在溶液状态下, 零价硅的π孤对电子离域于三个硅原子之间; 而在固体状态下, π孤对电子则定域在零价硅与其中一个硅宾配体之间. 反应化学研究表明, 化合物60可通过Si(0)原子转移反应生成氮杂环硅宾化合物.
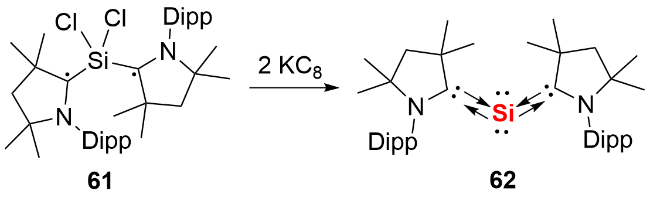
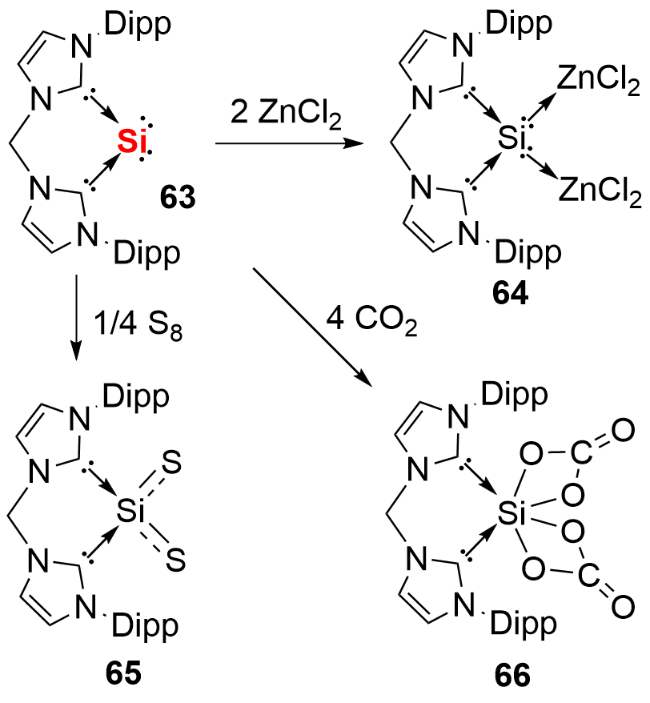
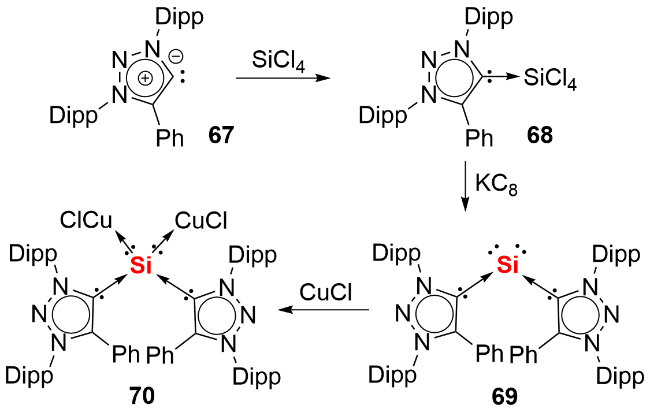
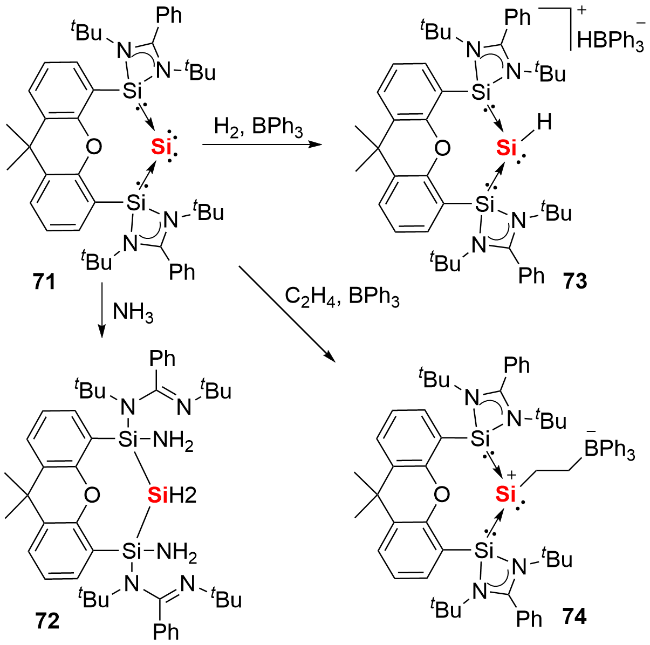
氯代脒基硅宾属于四元环状氮杂环硅宾化合物, 其环外的Si—Cl键可通过盐消除途径与各类取代基或分子骨架结合, 进而形成单齿或双齿硅宾. 相较于双齿卡宾, 双齿硅宾化合物的合成路线更为简便. 鉴于双齿配体的螯合效应在稳定低价元素中心方面的关键作用, 研究人员已开发出多种骨架的双齿硅宾配体, 并将其应用于零价元素化合物的合成中. 2019 年, Driess等[41]利用氧杂蒽双齿硅宾配体, 成功合成了环状单核零价硅化合物71(图式24). 具体合成步骤为: 首先通过双硅宾配体与二氯化硅的NHC配合物发生配体交换反应, 得到双硅宾配位的二价硅前体, 随后经KC8还原获得目标产物. 化合物71展现出丰富的反应活性: 不仅能与2 equiv. NH3反应生成1,3-二氨基取代的三硅烷衍生物72, 还能在BPh3存在下, 实现H2的活化裂解及乙烯的加成反应, 分别生成产物73和74.
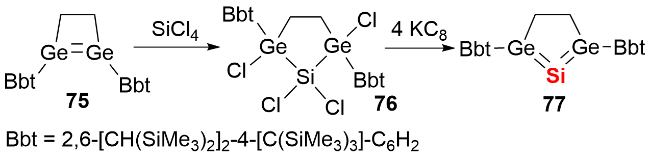
2008年, Robinson等[43]通过KC8还原SiCl₄的NHC配合物78, 以22%的产率分离得到首例NHC稳定的双核零价硅化合物79(图式26a). 单晶结构显示, 化合物79呈反式弯曲构型, Si—Si键长处于双键范围. 分子轨道计算表明, 其HOMO和HOMO-1分别对应硅硅π键和σ键, HOMO-2对应硅原子上的孤对电子, 每个Si原子均具有一对σ孤对电子. 采用类似合成路径, 2014年Roesky等[44]报道了CAAC稳定的双核零价硅化合物81的分离与表征(图式26b). 随着配体电子性质的改变, 双核硅化合物的结构特征呈现差异: 与化合物79相比, 化合物81的Si—C键长显著缩短, C—Si—Si键角增大, 这一现象与CAAC配体更强的π电子接受能力一致.
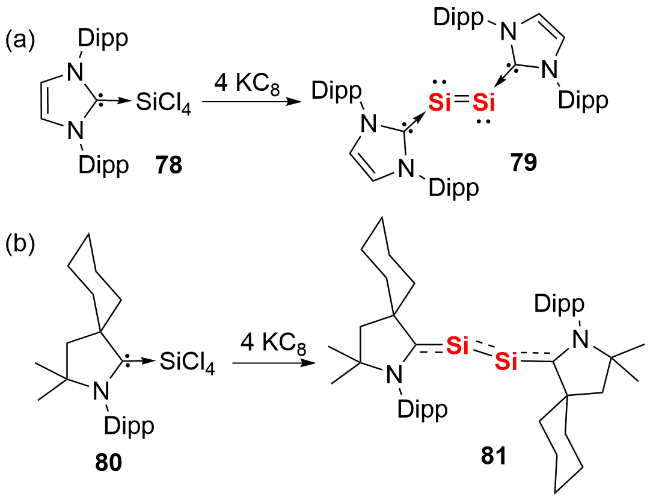
2022年, 我们[45]设计合成了兼具强给电子能力和大位阻效应的咪唑啉亚胺基硅宾配体82, 成功实现了硅宾稳定的双核零价硅化合物83的分离与表征(图式27). 通过将硅宾82、二氯化硅的NHC配合物与KC8进行“一锅法”反应, 以60%的产率获得目标化合物. 单晶结构分析表明, 化合物83的Si4片段呈反式弯曲构型, 中心Si—Si键长为0.2255(1) nm, 处于Si=Si双键的键长范围. 理论计算显示, 该Si—Si键由一个σ键(1.90 e⁻)和一个π键(1.80 e⁻)构成, 两个中心Si原子上各存在一对具有较高s型特征(71%)的孤对电子(1.88 e⁻), 完全符合双核零价硅的结构特征. 反应化学研究表明, 化合物83可与苯乙炔反应生成结构新颖的硅杂环化合物84, 凸显了其在构建复杂含硅化合物中的潜在应用价值.
4.3 零价锗化合物的合成及反应性
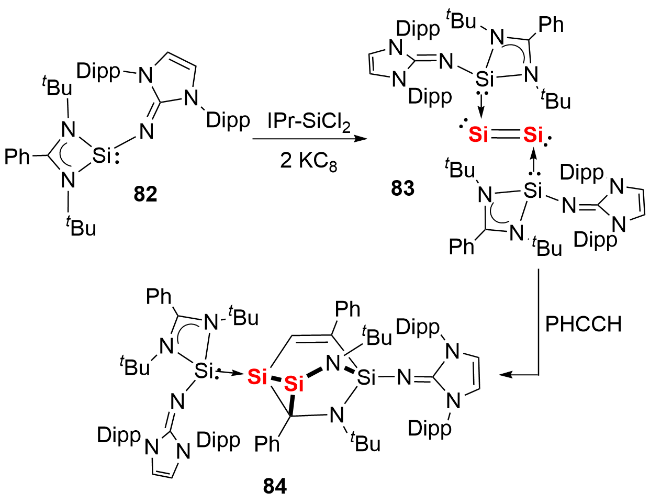
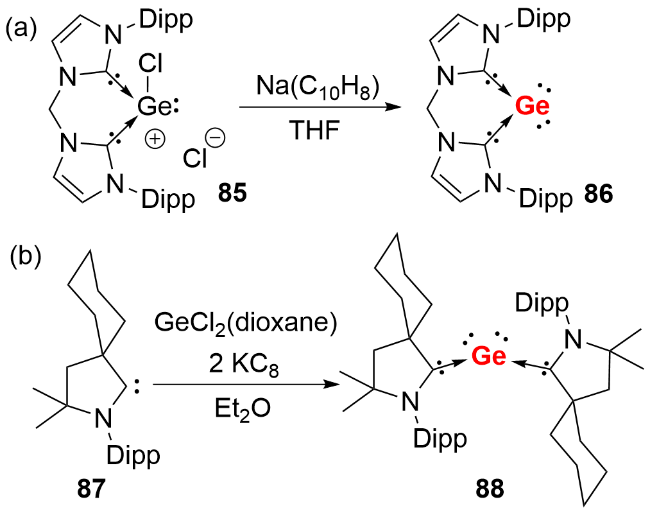
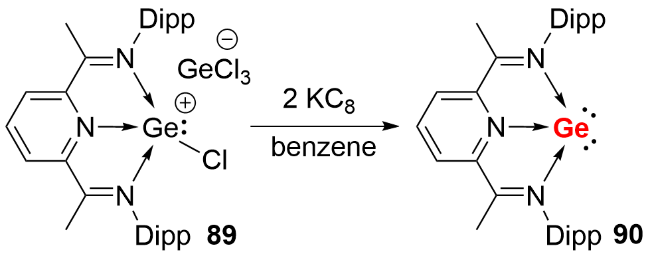
2013年, Driess等 [46]利用双齿卡宾配体成功合成了首例NHC稳定的单核零价锗化合物(germylones, 图式28a). 作者首先通过双齿卡宾配体与二价锗化合物GeCl2(dioxane)反应, 制备了卡宾配位的锗卤化物前体85, 随后经钠/萘还原得到目标化合物86. 单晶结构分析显示, 化合物86的Ge—C键长(0.1967(2) nm和0.1962(2) nm)处于单键范围, C—Ge—C键角为86.6(1)°. 理论计算表明, 其HOMO为锗原子的π型轨道, HOMO-1为锗原子的σ孤对电子轨道, 符合零价锗的结构特征. 同年, Roesky等[47]报道了CAAC稳定的单核零价锗化合物的分离与表征(图式28b). 作者以乙醚为溶剂, 在-78 ℃条件下将GeCl2(dioxane)、CAAC和KC8通过“一锅法”反应, 合成了深绿色目标化合物88. 同样由于CAAC配体更强的π电子接受能力, 其Ge—C键长(0.1954(2) nm和0.19386(18) nm)相比化合物86更短.
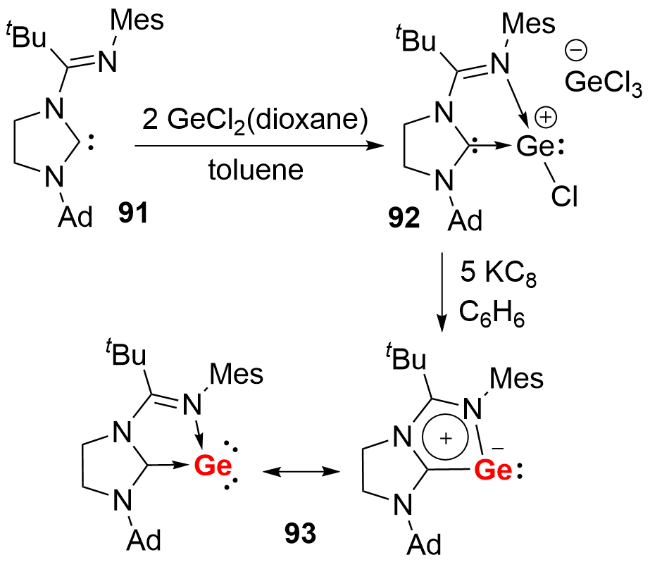
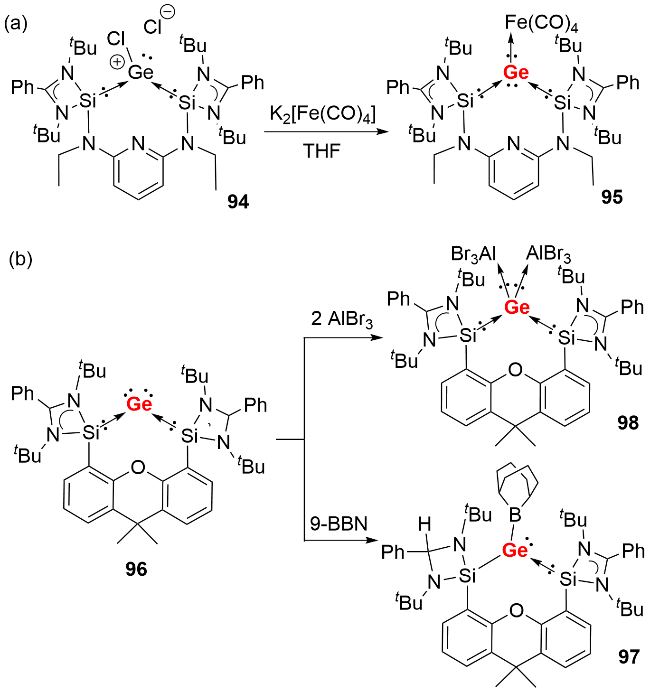
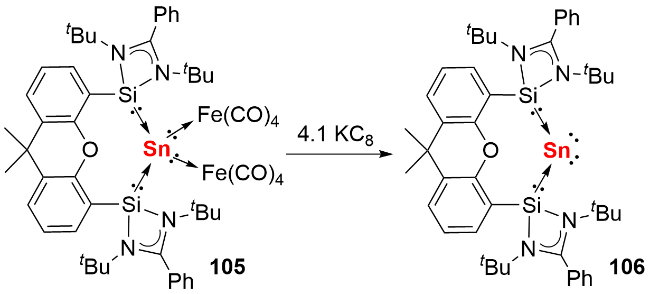
与零价硅化合物的合成策略类似, 双齿螯合硅宾配体同样适用于稳定单核零价锗化合物. 2016年, Driess 等[50]首先利用吡啶双硅宾配体合成了双硅宾配位的单核零价锗化合物95(图式31a), 其富电子的Ge(0)原子通过配位路易斯酸性的Fe(CO)4基团降低电子云密度, 从而实现结构稳定. 2021年, 该课题组进一步利用氧杂蒽双硅宾[51]和碳硼烷双硅宾[52]配体, 成功合成了系列单核零价锗化合物. 以氧杂蒽双硅宾稳定的化合物96为例, 其反应活性研究表明: 氧杂蒽双硅宾稳定的单核零价锗化合物96可以与2 equiv. AlBr3反应生成配合物98, 体现了锗中心两对孤对电子的配位能力; 还能活化9-硼双环[3.3.1]壬烷(9-BBN)的B—H键, 伴随氢迁移过程生成硼基-硅基锗宾化合物97(图式31b).
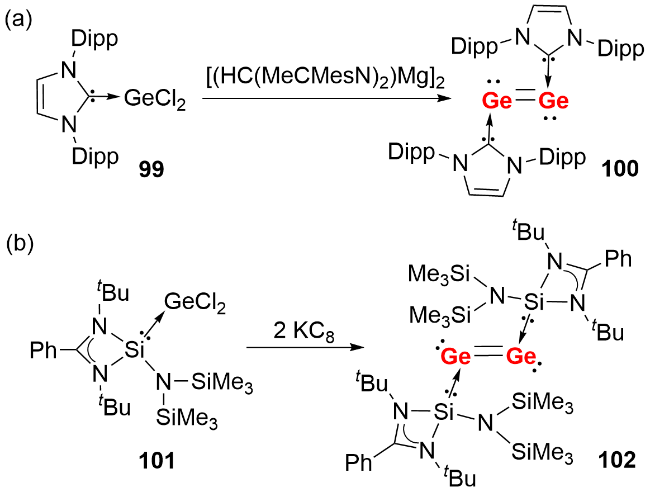
在双核零价锗化合物的合成研究中, 研究人员借鉴卡宾稳定双核零价硅的策略, 2009年Jones等[53]首先尝试采用KC8还原二氯化锗的NHC配合物制备双核零价锗, 未获成功; 随后将还原剂替换为一价镁试剂[(HC(MeCMesN)2)Mg]2, 最终以20%的产率分离得到NHC稳定的双核零价锗化合物100(图式32a). 该化合物呈反式弯曲构型, Ge—Ge键长为0.23490(8) nm, 处于双键键长范围. 2014年, So等[54]报道了氮杂环硅宾稳定的双核零价锗化合物的合成(图式32b), 其Ge—Ge键长为0.23518(16) nm, 同样属于双键范畴. 分子轨道计算表明, 其HOMO为Ge—Ge π键, 且该轨道与硅原子的空p轨道存在相互作用.
4.4 零价锡化合物的合成及反应性
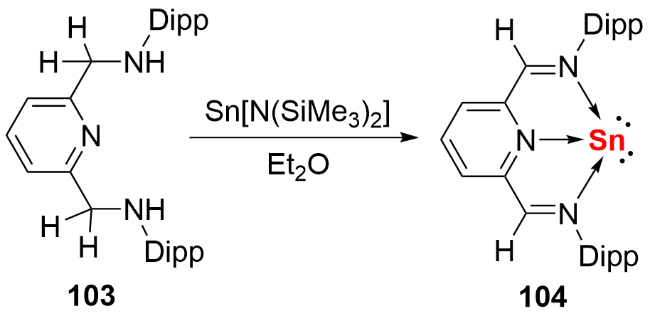
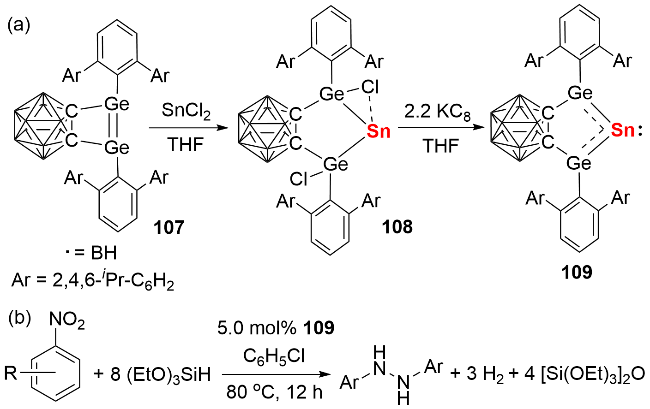
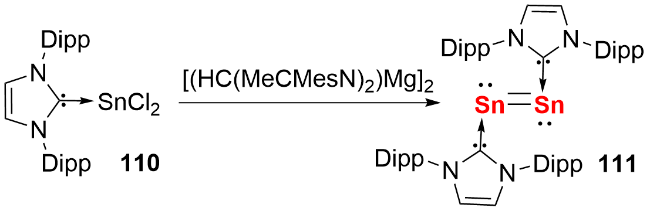
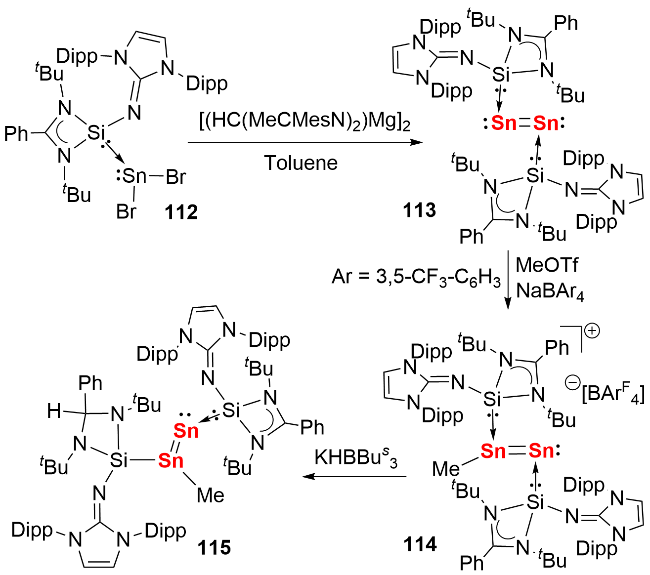
2023年, 我们[59]利用具有大位阻和强给电子能力的咪唑啉亚胺基硅宾配体, 成功构筑了硅宾稳定的双核零价锡化合物(图式37). 具体合成路径如下: 首先通过 SnBr2的卡宾配合物与硅宾配体发生取代反应, 以90% 的产率分离得到SnBr2的硅宾配合物112; 随后该中间体与还原剂[(HC(MeCMesN)2)Mg]2反应, 以60%的产率获得目标化合物113. 化合物113在室温甲苯溶液中表现出优异的稳定性, 数日内无分解迹象. 理论计算表明, 与传统卡宾体系相比, 其Sn—Sn键的σ键与π键电子向硅宾配体中Si原子空p轨道的反馈作用, 显著增强了分子结构的稳定性. 基于产率与稳定性的双重提升, 我们进一步探索了双核零价锡的反应化学性质: 通过甲基化反应制备了硅宾稳定的甲基锡阳离子化合物114, 该中间体再与三仲丁基硼氢化钾(KHBsBu3)反应, 成功合成了硅宾稳定的锡亚乙烯基化合物115.
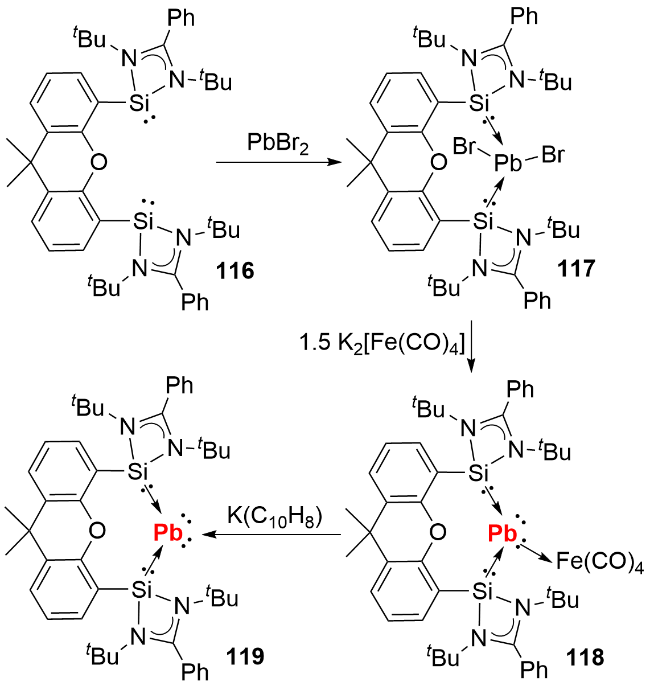
4.5 零价铅化合物的合成及反应性
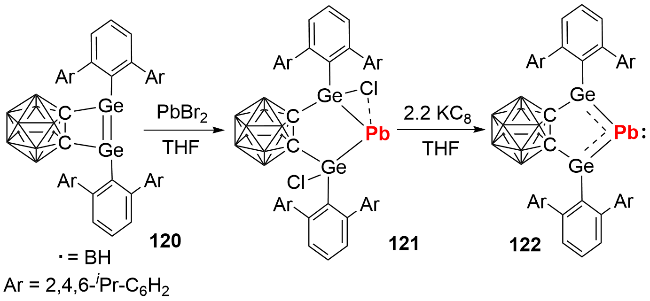
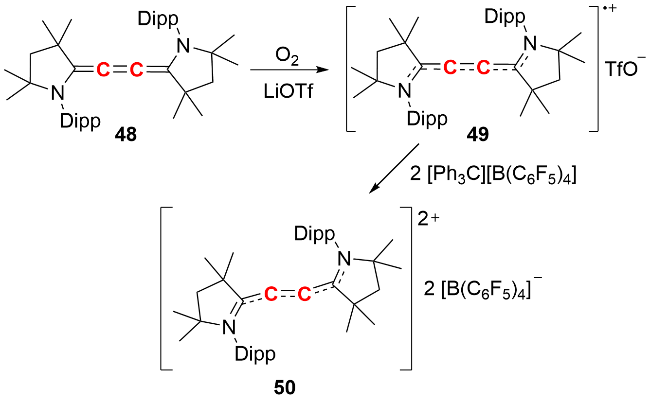
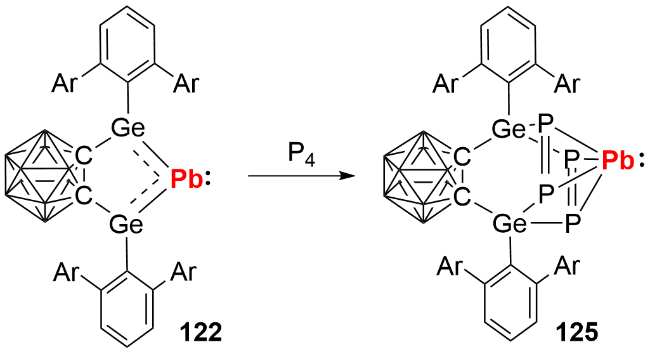
反应化学研究发现, 双锗宾零价铅化合物可与GeCl₂(dioxane)发生复分解反应生成双锗宾零价锗化合物123, 也能与芳基叠氮化合物发生铅原子转移反应生成产物124(图式40), 展现了其作为零价元素试剂在合成化学中的潜在应用价值.
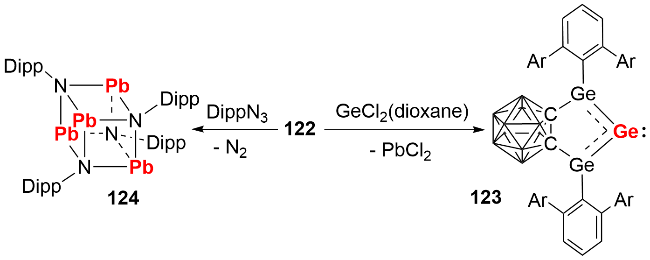
此外, 我们 [62]还通过双锗宾零价铅化合物与P4的反应, 成功构筑了双膦烯铅π配合物125(图式41). 晶体结构分析显示, 该化合物的P—P键长为0.2124(2) nm和0.2139(2) nm, 处于过渡金属膦烯π配合物的P=P双键键长范围(0.2081~0.2197 nm). 其核磁共振磷谱在δ -127.9处仅出现单峰, 表明结构中的四个P原子化学环境等价. 能量分解分析表明, 双膦烯配体对Pb(0)原子的σ给电子作用与Pb(0)原子向双膦烯的π反馈作用共同维持了化合物的稳定性, 其中π反馈作用占主要贡献. 该双膦烯铅π配合物的分离与表征, 为零价主族元素化合物的稳定机制提供了新思路.
5 零价态第15族元素化合物的合成及反应性
5.1 双核零价磷化合物的合成及反应性
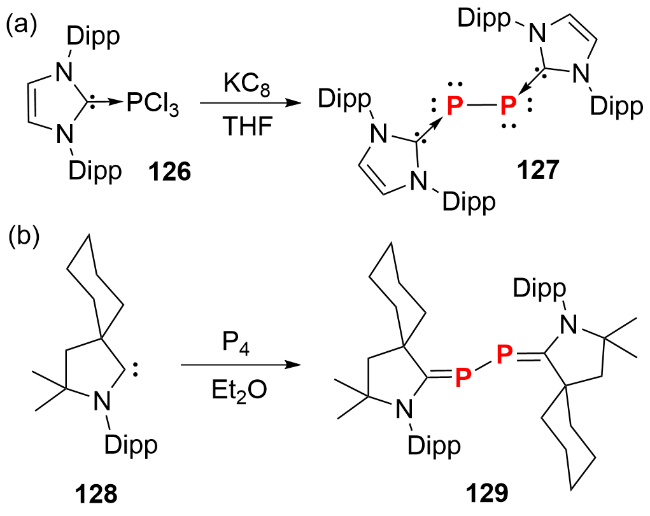
第15族元素中的氮与磷是构成生命活动的必需元素, 其单质(如氮气(N2)和白磷(P4))的活化及转化一直是化学研究的热点领域. 与N2不同, 磷元素难以以P2片段形式存在, 仅在800 ℃以上的高温及低压极端条件下, P4才会解离为P2片段. 因此, 稳定P2化合物的分离与表征, 可为含磷化合物的合成提供新的方法. 关于过渡金属促进P4活化生成P2化合物的研究已有报道. 除过渡金属外, 2008年Robinson等[63]通过KC8还原PCl3的NHC配合物126, 合成了形式上的双核零价磷化合物127(图式42a). 随后, Bertrand等[64]在CAAC配体直接活化P4的反应中, 分离出少量双核零价磷产物129(图式42b). 晶体结构显示, 两者的P—P键长分别为0.22052(10) nm和0.2184(3) nm, 均接近单键键长. 由于NHC和CAAC配体电子性质的差异, 后者亦可视为磷杂丁二烯类似物.
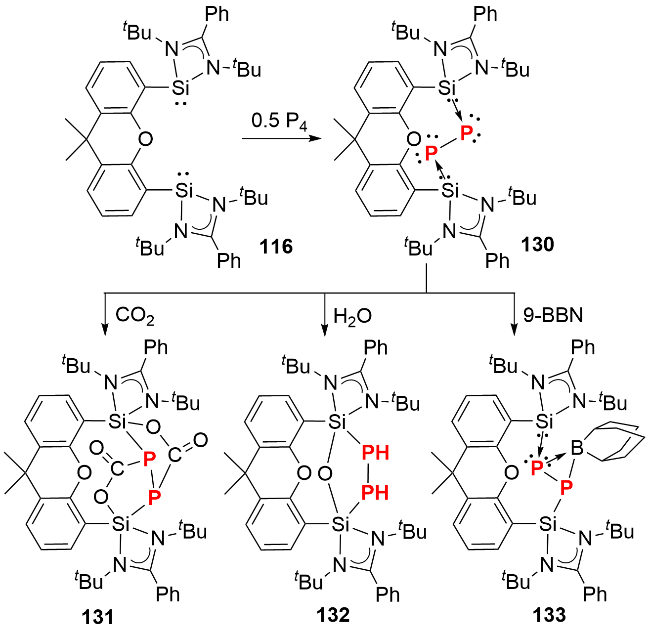
2020年, Driess等[65]利用氧杂蒽双硅宾配体与P4直接反应, 以91%的高产率合成了双硅宾稳定的双核零价磷化合物130(图式43). 该化合物中P—P键长为0.22369(8) nm, 处于单键键长范围; Si—P键长分别为0.21307(7)和0.21263(7) nm, 介于典型单键与双键之间. 自然键轨道分析表明, 其结构包含两个Si—P单键, 每个P原子携带两对孤对电子, 磷中心电荷为-0.59 a.u., 符合双核零价磷的结构特征. 与卡宾配体相比, 硅宾更强的σ给电子能力使其更接近电子供体-受体相互作用稳定的双核零价磷电子结构. 作者通过探究双核零价磷化合物与二氧化碳、水和9-BBN的反应, 成功实现了 P—C键、P—H键及P—B键的构建, 为含磷化合物的绿色合成提供了新的思路.
5.2 双核零价砷化合物的合成及反应性
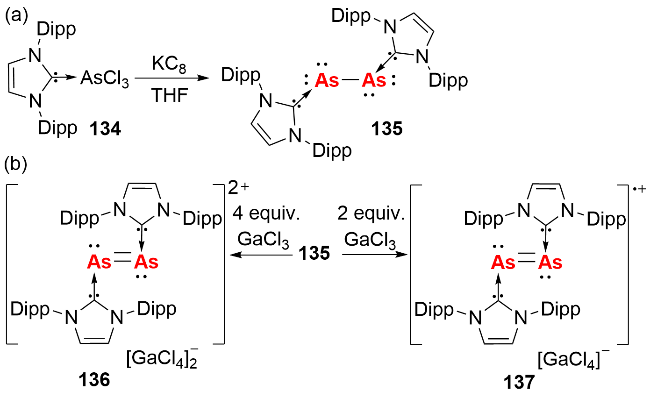
砷元素与磷元素性质相似, 其单质存在多种同素异形体, 常见的有灰砷、黄砷和黑砷. 其中黄砷(As4)与白磷分子结构类似, 由四个As原子以单键连接形成As4四面体结构, 且As4需在高温气相条件下才能分解为As2片段. 2009年, Robinson等[66]通过KC8还原AsCl3的NHC配合物, 成功合成了首例路易斯碱稳定的双核零价砷化合物135(图式44a). 该化合物中As原子间以单键结合, 每个As原子携带两对孤对电子. 其反应活性研究表明, 该双核零价砷化合物可以被4 equiv. GaCl3氧化, 生成含As=As双键的(As2)2+化合物136; 与2 equiv. GaCl3反应时, 则生成(As2)•+化合物137, 这是首例分离表征的砷自由基物种(图式44b).
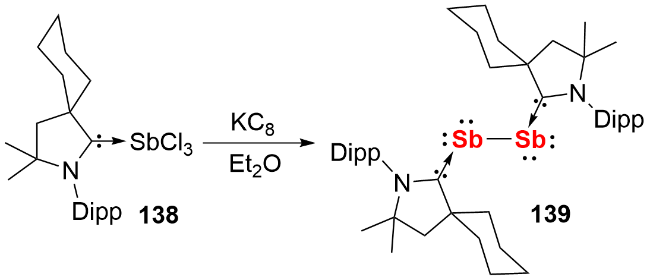
5.3 双核零价锑化合物的合成及反应性
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 总结与展望
零价主族元素化合物含有富电子的主族元素中心, 具有极高的反应活性, 长期以来难以实现稳定存在. 随着新型低价主族元素配体的设计与开发, 一系列具有开创性的零价主族元素化合物已被成功合成并分离表征. 由于配体电子性质的差异会显著影响零价元素原子与配体间的成键模式, 此类化合物通常展现出独特的电子结构特征及丰富多样的化学反应性. 本综述主要总结了元素周期表中p区第13至15族元素及少数s区碱土金属元素零价态化合物的合成, 阐述了其电子结构与成键方式, 介绍了其独特的化学反应性质, 旨在加深对零价态主族元素化合物的认识, 为更多此类化合物的开发与应用提供启示. 目前, 仍有诸多零价主族元素化合物尚未实现分离, 例如第13族中除硼以外的零价化合物、双核零价铅化合物、单核零价第15族元素化合物、双核零价铋化合物及碱土元素中相对较重的钙元素的零价化合物等. 稳定上述零价结构的关键在适配性配体的开发. 这些零价化合物的合成以及对其成键的研究, 将对后续低价主族元素配体的定向设计和高活性低价主族元素物种的稳定提供指导意义.
主族元素丰富的同素异形体在材料科学领域中一直扮演重要角色, 具有优异电学、热学及力学性能的新材料开发备受关注, 例如二维硼烯纳米材料等. 单核和双核零价主族元素化合物可作精准合成砌块, 结合其可控反应性, 有望制备新型功能材料, 如掺杂零价硅、磷的高效光电器件, 或基于零价硼的二维硼烯衍生物, 优化材料电学与力学等性能. 近期关于Ph3PCN2作为单原子C(0)转移试剂的研究, 展现了零价主族元素化合物在合成化学领域的广阔应用前景. 通过使用类似Ph3P与N2等易离去配体, 更多零价主族元素化合物作为零价元素试剂的应用值得期待. 此外, 在催化领域中, 依托零价元素中心与低价元素配体的协同作用, 构建低价主族元素催化体系具有重要的基础研究价值和潜在应用前景.
(Cheng, B.)