


1 引言
光化学反应凭借其温和的反应条件、良好的官能团兼容性以及广泛的底物适用性等显著优势, 已成为现代合成化学领域的研究热点[1]. 自2008年以来, 该领域呈现出爆发式增长, 相关研究呈指数级攀升. 当前主流的光催化可以分为三大类别: 光氧化还原催化[2]、氢原子转移催化[3]以及能量转移催化[4]. 尽管光催化的反应模式相对有限, 但其引发的自由基反应却展现出惊人的多样性. 根据自由基的电子特性, 可将其分为中性自由基与离子型自由基两大类别, 其中离子型自由基又可以进一步细分为阴离子型自由基和阳离子型自由基(Scheme 1). 经过十多年的发展, 基于光催化中性自由基参与的化学反应已取得丰硕成果[5], 与之形成鲜明对比的是, 离子型自由基的相关研究仍处于探索阶段. 现有研究多局限于零星报道[6-7], 尚未形成系统性的规模化研究.
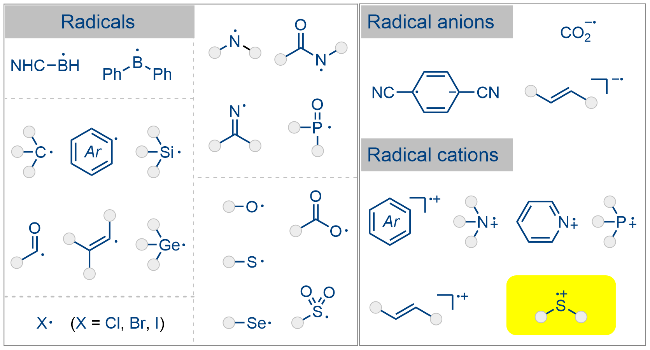
含硫化合物在光化学领域扮演着多重角色: 一方面可以作为高效有机光催化剂(如吩噻嗪类光催化剂)[8], 另一方面又可作为稳定的自由基前体参与到光化学反应中(如噻蒽盐及Umemoto试剂等)[9]. 值得注意的是, 对于上述自由基前体, 其体系中硫醚类化合物可能参与的后续光化学反应, 却鲜有研究. 本文以硫自由基阳离子为切入点, 系统阐述硫醚类试剂参与的光化学反应, 重点介绍此类自由基阳离子的产生方式、反应特性及底物适用范围. 根据硫自由基阳离子产生方式, 本综述分为以下三个部分: (1)光氧化还原催化产生硫自由基阳离子; (2)光催化S—X键均裂产生硫自由基阳离子; (3)吩噻嗪类光催化剂产生的硫自由基阳离子.
2 光催化产生硫自由基阳离子
2.1 光氧化还原催化产生硫自由基阳离子
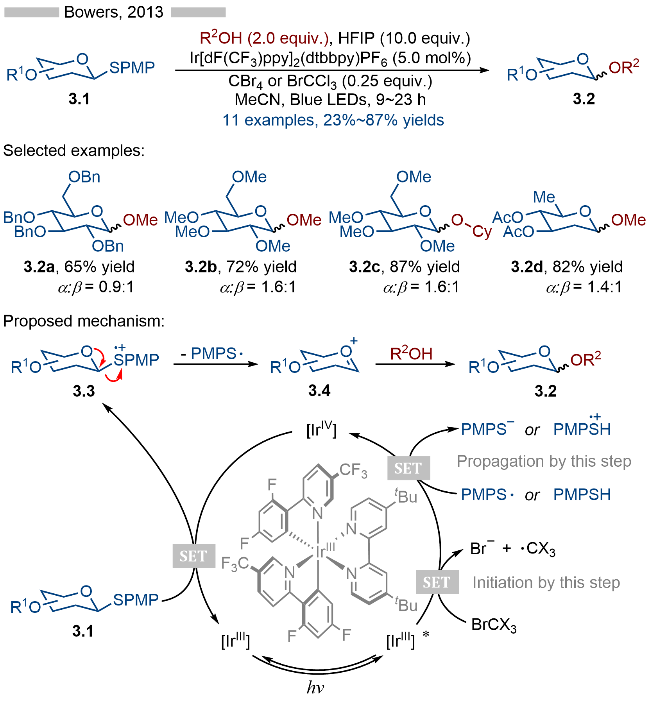
2013年, Bowers课题组[11]报道了硫代葡萄糖类化合物参与的光催化C—S键碎裂化/C—O键形成反应, 并以此实现了O-糖基化反应(Scheme 3). 该反应以强氧化性的Ir[dF(CF3)ppy]2(dtbbpy)PF6为光催化剂、以对甲氧基取代的苯基硫醚为离去基团、以卤代物为自由基引发剂. 在底物适用性方面, 无论是苄基、甲基还是乙酰基保护的底物都能很好地得到相应的目标产物. 其详细的反应机理如Scheme 3所示, 在光照条件下, 基态光催化剂变为激发态[IrIII]*, 随后激发态光催化剂与卤代物发生单电子转移(SET), 生成氧化态光催化剂[IrIV]. 氧化态光催化剂具有较强的单电子氧化能力, 其可以将底物3.1氧化为相应的硫自由基阳离子中间体3.3, 再经过C—S键碎裂化, 生成相应的氧鎓离子中间体3.4. 反应体系中的醇再与氧鎓离子中间体3.4发生亲核加成反应, 即可得到相应的O-糖基化产物3.2.
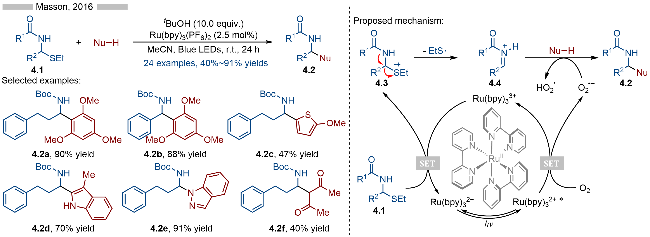
2016年, Masson课题组[12]报道了光催化α-硫代酰胺类化合物的C—S键碎裂化/C—X键形成反应, 并合成了一系列α,α-二取代酰胺类化合物(Scheme 4). 在光催化氧化条件下, α-硫代酰胺类化合物被氧化成相应的亚胺类中间体, 其再与各种亲核试剂反应, 生成相应的目标产物. 在底物兼容性方面, 无论是α-位烷基取代的底物(产物4.2a), 还是芳基取代的底物(产物4.2b)都能适用于该反应. 此外各种亲核试剂都能很好地参与该反应, 如均三甲氧基苯、噻吩、吲哚、吲唑以及1,3-二羰基类化合物等. 其详细的反应机理如Scheme 4所示, 首先是激发态光催化剂与氧气发生单电子转移, 生成氧化态光催化剂Ru(bpy)33+和超氧负离子中间体. 氧化态光催化剂(E1/2[Ru(bpy)33+/Ru(bpy)32+]=+1.29 V vs. SCE)具有较强的单电子氧化能力, 其可以将α-硫代酰胺类底物4.1[Eox=+1.17 V vs. SCE]氧化成相应的硫自由基阳离子4.3. 随后中间体4.3发生C—S键异裂, 生成相应的亚胺离子中间体4.4, 最后与亲核试剂发生加成反应, 生成相应的酰胺类化合物4.2. 反应体系中生成的超氧负离子中间体可以作为碱捕获体系中的质子.
2022年, 李子刚课题组[14]报道了光催化多肽类化合物的选择性烷基化反应, 实现了组氨酸类化合物的烷基化(Scheme 5). 该反应以硫代缩醛为相应的烷基化试剂, 通过光催化将其转化为相应的硫鎓离子中间体, 随后再与多肽上的组氨酸残基反应, 从而得到相应的产物. 该反应具有较高的官能团兼容性, 无论是酰胺、氨基、还是羧基都能适用于该反应. 此外含多种氨基酸片段的多肽类化合物, 也能特异性地识别其中的组氨酸结构. 通过该策略, 李子刚课题组实现了一系列多肽类化合物的选择性烷基化反应, 其中包括亮丙瑞林(Leuprorelin, 产物5.3b)、血管紧张素II(Angiotensin II, 产物5.3c)以及美拉诺坦(Melanotan-I, 产物5.3d)等. 该反应的详细机理如Scheme 5所示, 首先有机光催化剂—孟加拉红(RB)在光照条件下, 生成激发态光催化剂RB*. 硫代缩醛5.1经过激发态光催化剂氧化, 生成相应的硫自由基阳离子中间体5.4, 再经过C—S键断裂, 即可得到相应的硫鎓离子中间体5.5. 中间体5.5可以选择性地与组氨酸上的咪唑发生亲核加成反应, 最终生成相应的硫醚类化合物5.3. 反应中的还原态光催化剂则是被空气氧化, 以此实现其催化循环.
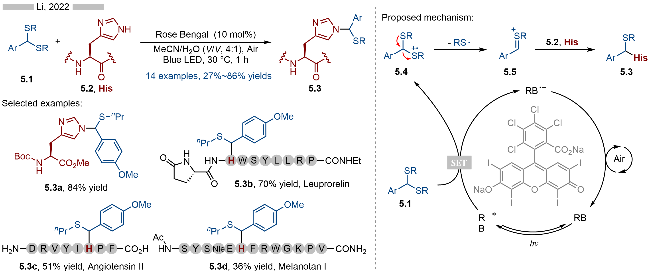
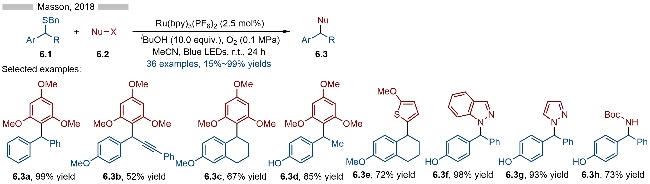
2018年, Masson课题组[15]报道了光催化苄基硫醚类化合物的选择性C—S键活化反应(Scheme 6). 该反应通过光催化氧化, 生成相应的硫自由基阳离子中间体, 再经过C—S键碎裂化, 生成相应的苄基碳正离子中间体, 最后再与各种亲核试剂反应, 即可得到相应的目标产物6.3. 反应具有较高的官能团兼容性和底物适用性, 无论是炔基、甲氧基还是酚羟基都能很好地适用于该反应. 在亲核试剂的选择方面, 均三甲氧基苯(产物6.3a~6.3d)、噻吩(产物6.3e)、吲唑(产物6.3f)、吡唑(产物6.3g)以及烯胺类化合物(产物6.3h)都能很好地参与到该反应当中. 此外, 作者还实现了基于光催化氧气直接氧化的苄位C—S键碎裂化, 该反应无需添加额外的光催化剂. 不过该策略的底物普适性相对较差, 只能实现部分底物的转化.
总的来说, 通过光催化氧化生成的碳正离子中间体, 可以实现氧α-位、氮α-位、硫α-位以及苄位的C—S键的碎裂化.
此外硫自由基阳离子也可以像氮自由基阳离子一样, 实现其α-位C—H键的碎裂化. 2020年, Hande课题组[16]报道了光催化与Brønsted碱共催化的硫醚类化合物的α-位烷基化反应(Scheme 7A). 无论是芳基烷基硫醚还是二烷基硫醚都能适用于该反应. 此外, 1,4-噻噁烷类化合物(产物7.3g)也能选择性地活化其中的硫α-位C—H键. 其详细的反应机理如Scheme 7A所示, 首先基态有机光催化剂Mes-tBu-Acr+吸收可见光, 生成激发态光催化剂Mes-tBu-Acr+*. 激发态光催化剂具有极强的单电子氧化能力, 其可以将硫醚类底物7.1氧化为相应的硫自由基阳离子中间体7.4. 硫自由基阳离子可以有效增强其α-位C—H键的酸性, 随后在Brønsted碱催化下, 中间体7.4失去质子, 生成硫α-位烷基自由基7.5. 该过程与氮自由基阳离子的α-位C—H键活化类似. 生成的烷基自由基7.5可以与缺电子烯烃发生自由基加成反应, 生成相应的羰基α-位烷基自由基, 再经过单电子还原/质子化等过程, 即可得到相应的烷基化产物7.3. 反应体系中生成的三氟乙酸可以与缺电子烯烃上的羰基部分结合, 以此提高烯烃的反应活性.
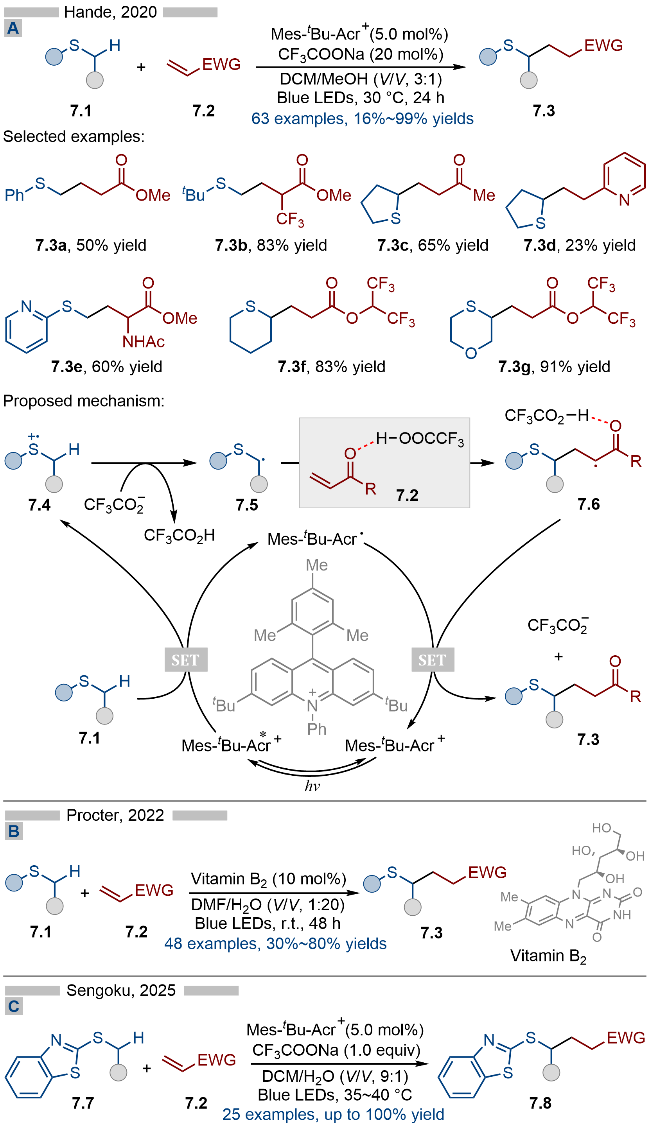
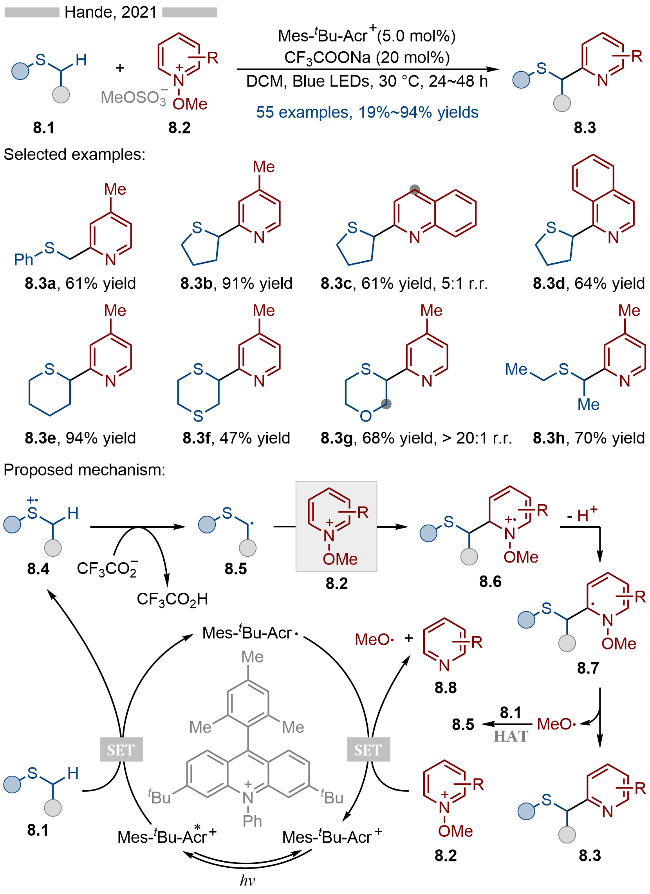
2021年, Hande课题组[19]报道了光催化和Brønsted碱共催化的硫醚类化合物的α-位杂芳基化反应(Scheme 8). 该反应以常见的Mes-tBu-Acr+为光催化剂, 以催化量的三氟乙酸钠作为Brønsted碱催化剂, 实现了硫醚的α-位C—H键的碎裂化. 在底物适用性方面, 无论是芳基烷基硫醚(产物8.3a)、环状硫醚(产物8.3b~8.3g)还是链状的二烷基硫醚(产物8.3h)都能兼容于该反应. 此外含多个活性位点的1,4-噻噁烷类化合物也能以极高的区域选择性, 得到相应的α-位芳基化产物8.3g. 在机理研究方面, 作者通过动力学同位素实验证明了硫α-位的C—H碎裂过程为决速步. 其详细的反应机理如Scheme 8所示, 首先激发态光催化剂Mes-tBu-Acr+*与硫醚类底物8.1发生单电子转移, 生成还原态的光催化剂Mes-tBu-Acr•以及硫自由基阳离子中间体8.4. 随后该自由基阳离子中间体在三氟乙酸根存在下去质子化, 生成相应的硫α-位烷基自由基8.5. 烷基自由基8.5与N-甲氧基杂芳烃盐类底物8.2发生Minisci类型反应, 生成氮自由基阳离子中间体8.6, 再经过去质子化/N—O键碎裂化, 即可得到相应的目标产物. 此外还原态光催化剂与底物8.2发生单电子转移, 生成基态光催化剂、氮杂芳烃8.8和甲氧基自由基. 反应体系中生成的甲氧基自由基可以与底物发生氢原子转移反应(HAT), 从而实现硫的α-位C—H键活化. 作者通过机理实验证明了该氢原子转移过程的存在.
2023年, Scheidt课题组[20]报道了光催化硫醚类化合物的α-位酰基化反应, 并合成了一系列β-酰基取代的硫醚类化合物(Scheme 9). 该反应以酰基取代的咪唑盐类化合物为相应的酰基化试剂, 通过光催化方式, 实现其与硫醚类底物的自由基-自由基偶联反应. 在底物适用性方面, 无论是芳基取代的酰基类底物(产物9.3a和9.3b), 还是烷基取代的酰基类底物(产物9.3c和9.3d)都能较好地适用于该反应. 此外无论是链状硫醚(产物9.3a~9.3f), 还是环状硫醚(产物9.3g和9.3h)也都能很好地适用于该反应. 其详细的反应机理如Scheme 9所示, 首先是激发态的光催化剂4CzIPN*与酰基咪唑盐类底物9.2发生单电子转移反应, 生成氧化态光催化剂和咪唑盐类自由基中间体9.4. 与此同时, 硫醚类底物被氧化态光催化剂氧化成相应的硫自由基阳离子中间体9.5, 再经过去质子化, 即可得到相应的硫α-位烷基自由基9.6. 烷基自由基9.6可以与咪唑盐类自由基中间体9.4发生自由基-自由基偶联反应, 生成相应的含咪唑盐类结构的醇类化合物9.7. 该中间体是通过核磁和X-射线单晶衍射等手段得以确定. 在1,8-二氮杂二环十一碳- 7-烯(DBU)存在下, 化合物9.7发生C—C键碎裂化, 生成相应的β-酰基取代的硫醚类化合物9.3.
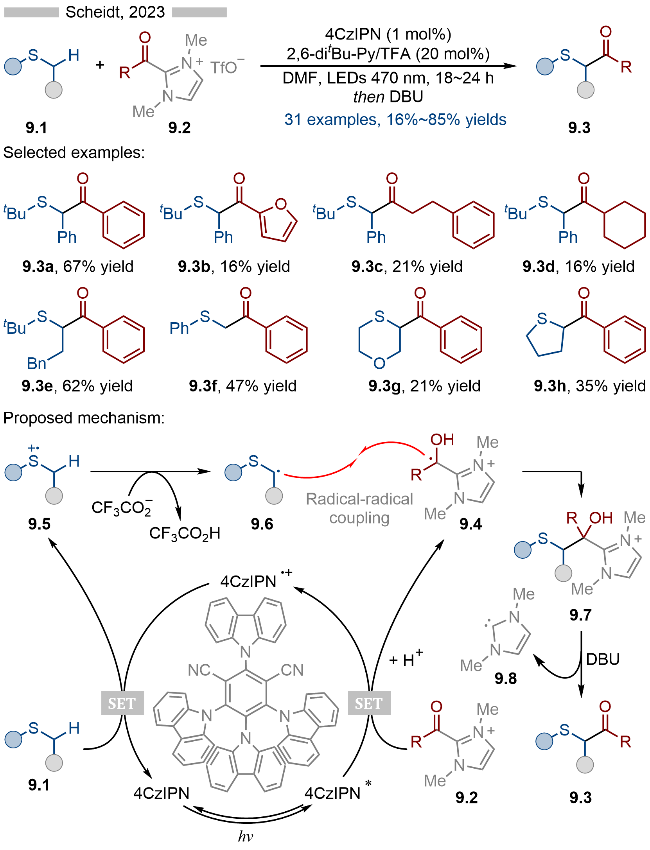
2023年, 龚磊课题组[21]报道了光催化芳基硫醚类化合物的自由基环化反应, 并合成了一系列多元杂环类化合物(Scheme 10). 该反应以芳基烷基硫醚为相应的烷基自由基前体, 以马来酰亚胺为相应的自由基受体, 通过串联环化反应, 实现了苯并噻喃类结构的合成. 在底物适用性方面, 马来酰亚胺的氮原子上无论是芳基(产物10.3a和10.3b)、甲基(产物10.3c)还是氢原子(产物10.3d)都能适用于该反应. 其详细的反应机理如Scheme 10所示, 首先是硫醚类底物10.1被激发态光催化剂氧化成相应的硫自由基阳离子10.4, 随后在硫酸根存在下去质子化, 生成相应的硫α-位烷基自由基10.5. 接着烷基自由基10.5与马来酰亚胺类底物10.2发生自由基加成反应, 生成相应的羰基α-位烷基自由基10.6. 自由基10.6再经过分子内的自由基环化/单电子氧化/去质子化等过程, 即可得到相应的苯并噻喃类化合物10.3. 此外烷基自由基10.6也有可能被氧化为相应的碳正离子中间体10.8, 随后再经过分子内的Friedal-Crafts烷基化反应, 也能得到相应的目标产物.
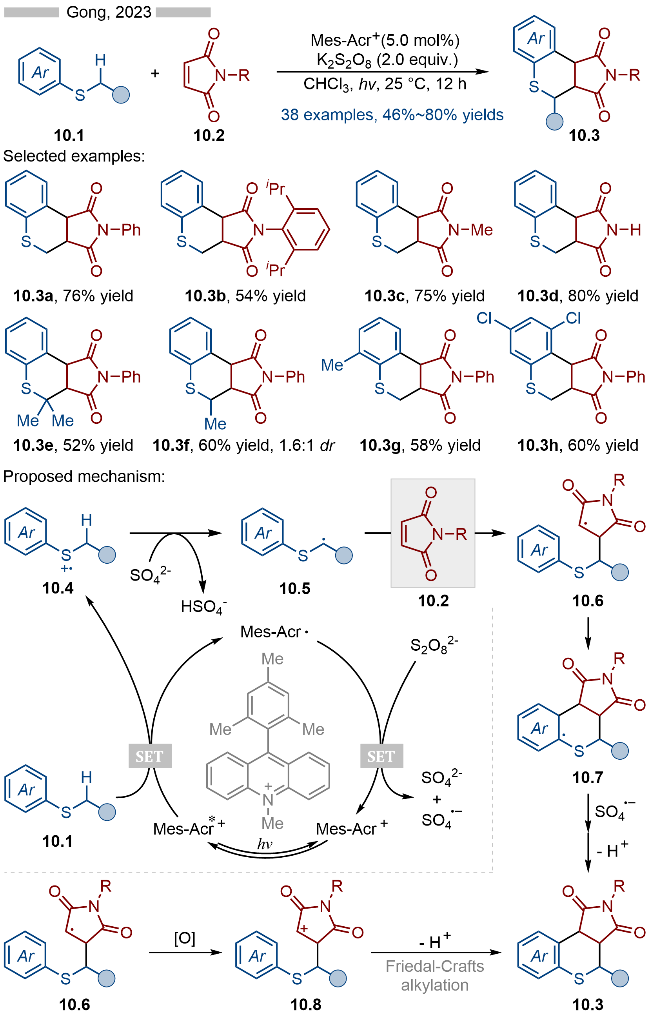
2024年, 赵玉龙课题组[22]报道了基于光催化硫醚参与的异氰类化合物的自由基环化串联反应, 并合成了一系列2-硫代喹啉类化合物(Scheme 11). 该反应以硫醚作为自由基阳离子前体, 通过光催化自由基加成/环化/异构化等过程, 将邻位乙烯基取代的芳基异氰转化成喹啉类化合物. 当底物为甲基叔丁基硫醚或甲基苄基硫醚时, 其产物结构上则优先保留甲基部分(产物11.3a). 当甲基取代硫醚结构的另一侧是乙基(产物11.3c)、正丙基(产物11.3d)或芳基(产物11.3e)时, 则优先消除其中的甲基部分. 此外二苯基硫醚也能适用于该反应, 并得到相应的目标产物11.3b. 其详细的反应机理如Scheme 11所示, 首先激发态光催化剂与DBU发生单电子转移, 生成DBU自由基阳离子, 该自由基阳离子可以与硫醚11.1发生单电子转移, 生成相应的硫自由基阳离子中间体11.4. 其中的DBU则是起到了电子转移催化剂的作用. 生成的硫自由基阳离子11.4可以与异氰发生自由基加成反应, 生成亚胺自由基阳离子11.5. 中间体11.5发生分子内的自由基环化, 生成自由基中间体11.6, 再经过分子内环化/异构化, 得到相应的阳离子中间体11.8. 上述过程中失去的溴自由基可以被还原态光催化剂还原为溴负离子, 随后溴负离子再与11.8反应, 即可得到相应的2-硫代喹啉类化合物11.3.
总的来说, 硫醚类化合物经过光催化氧化后, 可以产生相应的硫自由基阳离子, 其既可以实现C—S键碎裂化, 又可以促使其α-位发生C—H键活化. 不过基于硫自由基阳离子参与的催化反应却很少受到合成化学家的关注, 相关研究也比较滞后.
2019年, Ritter课题组[23]报道了光催化芳基噻蒽盐类化合物的氟化反应, 并合成了一系列氟代芳烃类化合物(Scheme 12A). 该反应经历两步过程, 实现了芳基C(sp2)—H键向C(sp2)—F键的转化. 首先将芳烃类底物转化为相应的噻蒽盐类化合物, 再通过光催化实现C—S键碎裂化/C—F键偶联等过程, 从而实现芳烃的氟化反应过程. 反应体系中需要加入大量的一价铜盐作为其偶联催化剂, 而氟负离子则来源于CsF. 该反应具有良好的官能团兼容性, 无论是酚羟基(产物12.2b)、醇羟基(产物12.2c)、醛基(产物12.2d)还是酰胺基团(产物12.2e)都能适用于该反应. 其详细的反应机理如Scheme 12A所示, 首先激发态光催化剂[IrIII]*将反应体系中游离的噻蒽氧化为噻蒽自由基阳离子, 随后还原态光催化剂[IrII]与噻蒽盐类底物发生单电子转移, 生成基态光催化剂和芳基自由基12.3. 另一方面, 一价铜[CuIF]被噻蒽自由基阳离子氧化成二价铜[CuIIF2], 随后其与芳基自由基12.3结合, 生成三价铜中间体[ArCuIIIF2], 再经过还原消除, 即可得到C—F键偶联产物12.2. 反应体系中的噻蒽起到了电子转移催化剂的作用.
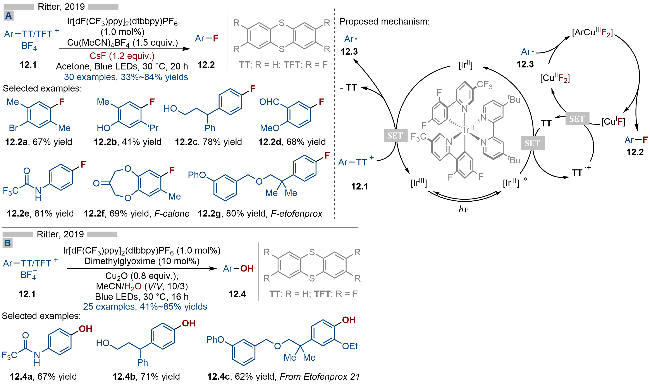
同年, 基于相同的反应策略, Ritter课题组[24]报道了光催化的噻蒽盐类化合物的羟基化反应, 并合成了一系列酚类产物(Scheme 12B). 该反应同样经历了两步反应过程, 首先是将芳烃类化合物转化为相应的噻蒽盐, 实现了芳基C(sp2)—H的活化, 再通过光催化C—O键偶联, 将其转化为相应的酚类产物. 产物结构上的酚羟基来源于水. 反应体系中的噻蒽同样是起到了电子转移催化剂的作用.
2022年, 吴雪松课题组[25]报道了光催化和噻蒽共催化醇的β-位碎裂化反应(Scheme 13). 该反应通过协同催化产生烷氧自由基, 实现其β-位C—C键的碎裂化. 在底物拓展方面, 无论是苄醇(13.1a, 13.1g和13.1h)、二级醇(13.1b和13.1d)、一级醇(13.1c)、还是三级醇(13.1e和13.1f)都能很好地兼容于该反应, 实现其β-位碎裂化反应. 其详细的反应机理如Scheme 13所示, 首先激发态光催化剂4CzIPN*与噻蒽发生单电子转移, 生成还原态光催化剂和噻蒽自由基阳离子. 随后噻蒽自由基阳离子与羟基结合, 生成相应的烷氧基取代的噻蒽盐类中间体13.4. 中间体13.4再经过光催化单电子还原/O—S键碎裂化, 生成相应的烷氧自由基13.5. 烷氧自由基很容易发生β-裂解, 生成相应的烷基自由基13.6. 烷基自由基再经过自由基加成/单电子还原/质子化等过程, 即可得到相应的目标产物13.3.
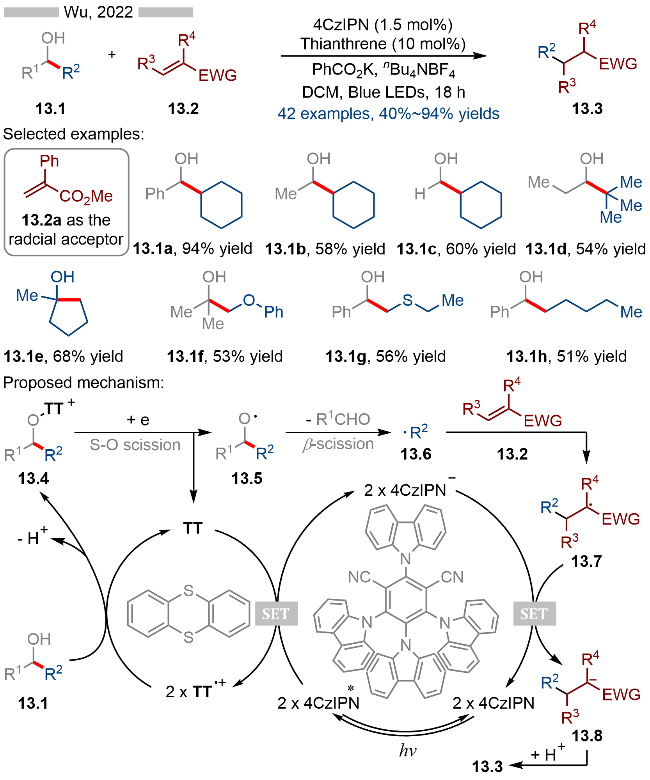
2023年, 吴雪松课题组[26]基于前期的研究基础, 实现了光/噻蒽/甲醇三组分协同催化的C(sp3)—H官能团化反应, 实现了一系列惰性碳氢键的烷基化(Scheme 14). 当反应中存在多种C(sp3)—H键时, 其存在一定的区域选择性(产物14.1b和14.1c). 其反应机理与Scheme 13类似, 光催化氧化产生的噻蒽自由基阳离子可以与甲醇结合, 从而生成相应的甲氧基取代的噻蒽盐类中间体14.4. 中间体14.4再经过光催化单电子还原, 即可发生O—S键碎裂化, 生成相应的甲氧基自由基14.5. 高活性的甲氧基自由基可以与烷烃类底物发生氢原子转移反应, 重新生成甲醇, 从而实现甲醇的催化循环. 烷烃经过氢原子转移反应后, 则可以转化为相应的烷基自由基14.6, 随后再经过分子间的自由基加成/单电子还原/质子化等过程, 即可得到相应的烷基化产物14.3.
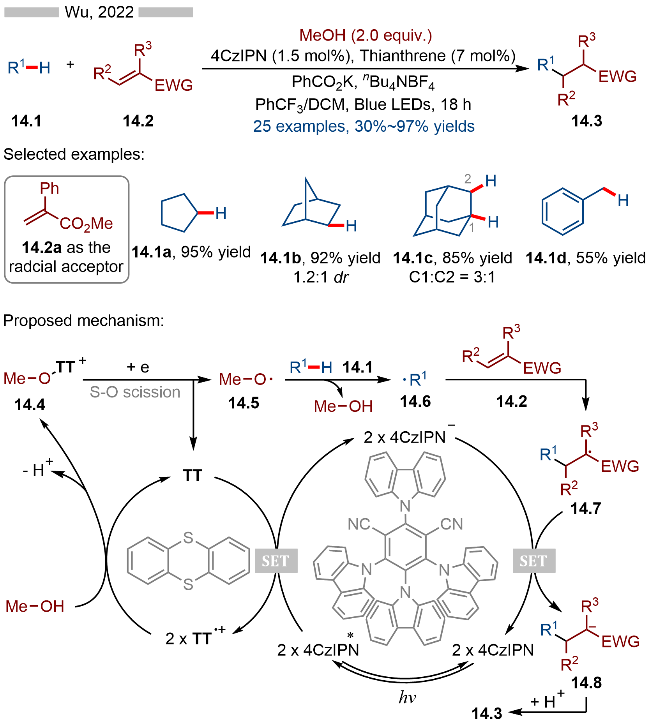
2023年, Saito课题组[27]报道了光催化和硫醚催化相结合的协同催化反应, 实现了烯烃的氯三氟甲基化(Scheme 15). 该反应以三氟甲磺酰氯为三氟甲基源和氯源, 以Ru(bpz)3(PF6)2为光催化剂, 二异丙基硫醚为共催化剂, 非活性烯烃为自由基受体. 当反应体系中没有二异丙基硫醚时, 反应也能发生, 但产率相对较低(15.3g和15.3h). 其详细的反应机理如Scheme 15所示, 首先, 激发态的光催化剂Ru(bpz)3(PF6)2*具有一定的单电子还原能力, 其可以将三氟甲磺酰氯还原为相应的三氟甲磺酰基自由基15.4, 再经过脱二氧化硫过程, 即可转化成相应的三氟甲基自由基15.5. 亲电性的三氟甲基自由基可以与烯烃发生加成反应, 生成烷基自由基15.6. 另一方面氧化态光催化剂Ru(bpz)33+可以将二异丙基硫醚氧化成相应的硫自由基阳离子, 其与15.6发生自由基-自由基偶联反应, 生成相应的硫鎓离子中间体15.7. 反应体系中的氯负离子随后与15.7反应, 即可得到相应的目标产物15.3. 如果反应体系中没有硫醚的话, 烷基自由基15.6则会被氧化成相应的碳正离子中间体15.8, 随后再与氯负离子反应. 当反应体系中存在硫醚时, 其生成的硫自由基阳离子可以有效捕获烷基自由基15.6, 以此形成相对稳定的硫鎓离子中间体.
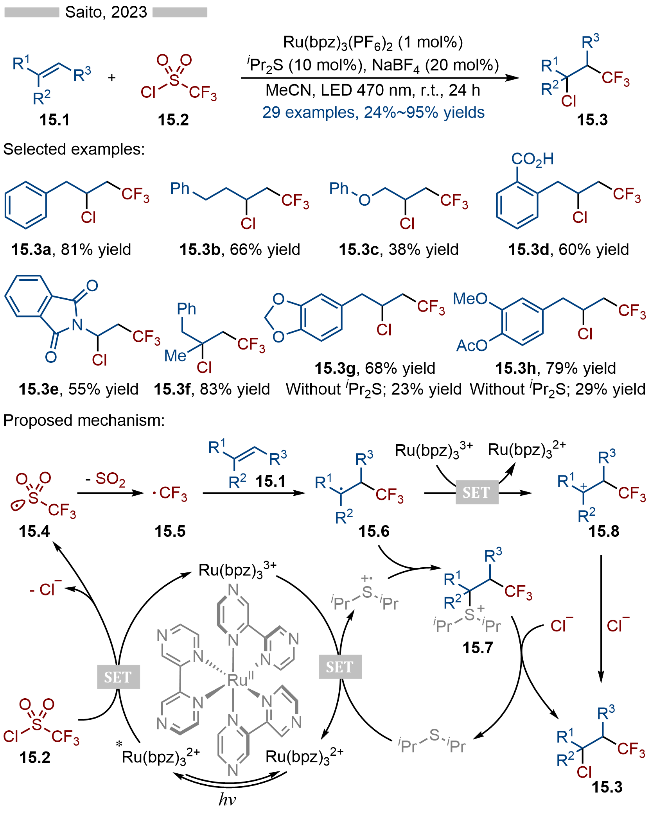
随后在2025年, Saito课题组[28]报道了光/硫醚共催化的多组分串联反应, 实现了烯烃的氨基-烷基化(Scheme 16). 该反应以邻苯二甲酰亚胺衍生的羧酸酯类化合物为烷基化试剂, 腈类溶剂为相应的胺源, 二异丙基硫醚为相应的催化剂. 该反应具有良好的官能团兼容性, 无论是一级烷基羧酸(产物16.4a)、二级烷基羧酸(产物16.4b)还是三级烷基羧酸(产物16.4c)衍生的活性酯都能很好地适用于此反应. 此外缺电子的肉桂酸酯类底物也能够有效参与该反应, 并以此合成相应的β-氨基酸酯类化合物16.4d. 其详细的反应机理如Scheme 16所示, 首先激发态光催化剂[IrIII]*与二异丙基硫醚发生单电子转移, 生成相应的还原态光催化剂[IrII]和硫自由基阳离子中间体. 活性酯类底物16.1经过还原态光催化剂[IrII]还原, 发生N—O键/C—C键碎裂化, 生成相应的烷基自由基16.5. 烷基自由基16.5与苯乙烯类型底物16.2发生自由基加成反应, 生成相应的苄基自由基16.6. 苄基自由基16.6与硫自由基阳离子发生自由基-自由基偶联反应, 生成相应的硫鎓离子中间体16.7, 再经过C—S键碎裂化, 即可生成相应的苄基碳正离子中间体16.8. 碳正离子中间体与溶剂中的氰基部分结合, 生成相应的氮正离子中间体16.9, 随后再经过亲核加成/重排等过程, 即可得到相应的酰胺类化合物16.4.
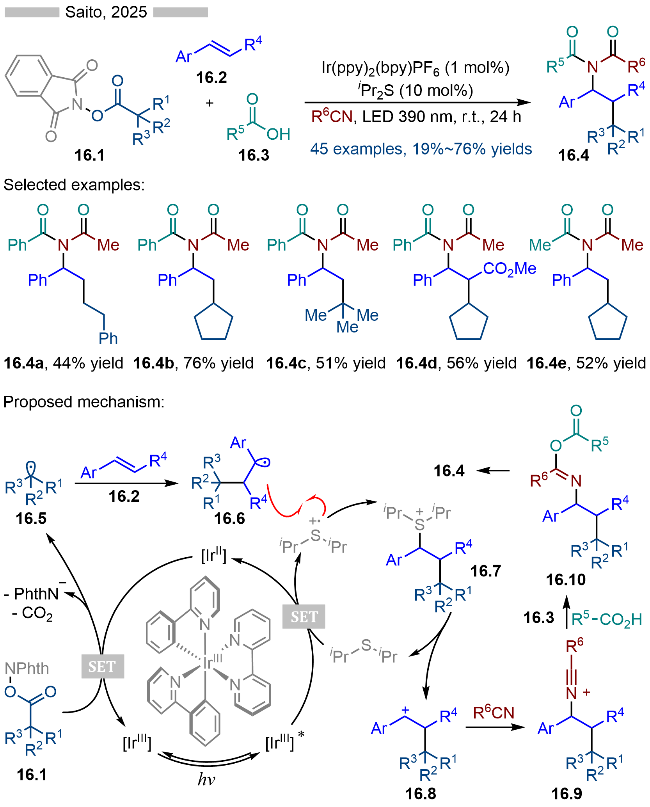
同年, Saito课题组[29]又报道了光/硫醚共催化烯烃的氧化-烷基化反应, 并合成了一系列多取代四氢呋喃类化合物(Scheme 17). 该反应以β-羟基酸衍生的活性酯为相应的烷基自由基前体, 其中的羟基部分为结构上的亲核位点, 从而实现后续的分子内环化过程. 经过底物设计, 作者合成了一系列螺环类化合物. 其详细的反应机理如Scheme 17所示, 首先是激发态光催化剂与二异丙基硫醚发生单电子转移, 生成相应的还原态光催化剂和硫自由基阳离子. 还原态光催化剂与活性酯发生单电子转移反应, 生成基态光催化剂和烷基自由基中间体17.4. 烷基自由基17.4与苯乙烯类型底物发生自由基加成反应, 生成相应的苄基自由基17.5. 苄基自由基17.5与硫自由基阳离子发生自由基-自由基偶联反应, 生成相应的硫鎓离子中间体17.6. 作者通过密度泛函理论(DFT)计算表明硫鎓离子中间体17.6很容易发生C—S键碎裂化, 生成相应的苄基碳正离子中间体17.7, 实现形式上的苄基自由基的单电子氧化. 碳正离子中间体17.7可以与分子内的羟基发生SN1反应, 生成相应的多取代四氢呋喃类化合物17.3.
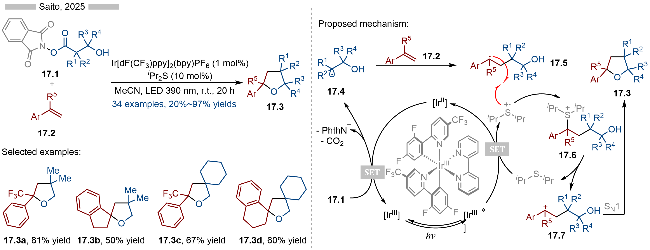
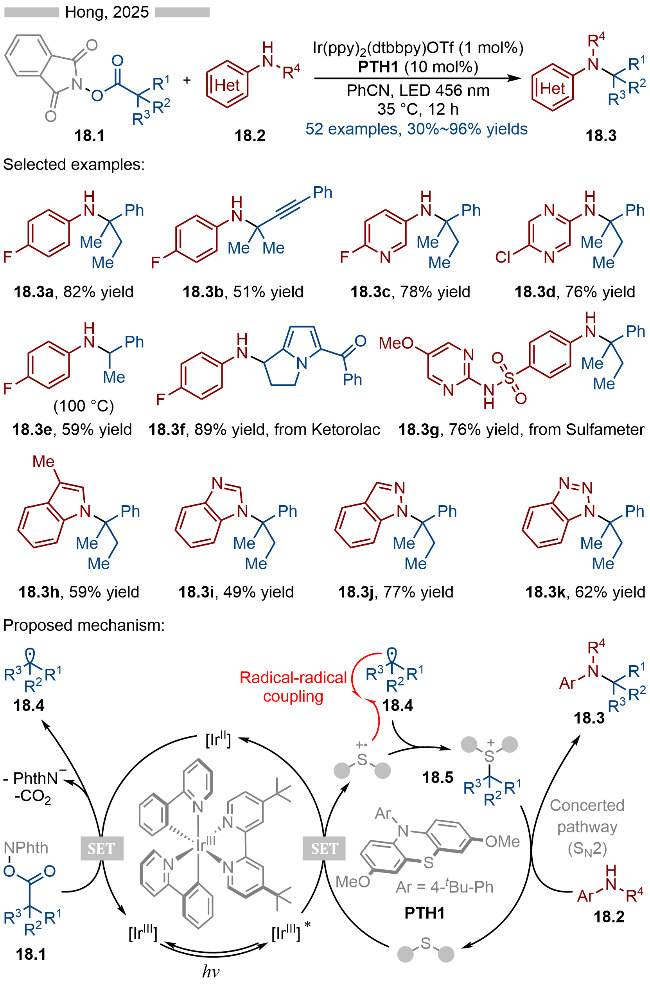
2025年, Hong课题组[30]报道了光/硫醚协同催化的C—N键偶联反应, 实现了羧酸类衍生物的脱羧氨基化 (Scheme 18). 在底物适用性方面, 各种富电子的芳基胺类化合物以及含氮杂环类化合物都能参与到该反应中. 在机理研究方面, 作者测定了催化剂PTH1的氧化电位为E1/2ox=0.40 V vs. SCE, 苯胺的氧化电位为Ecalcox=0.96 V vs. SCE, 产物18.3的氧化电位为Ep/2=0.88 V vs. SCE. 因此, 硫醚类催化剂的存在, 有效避免富电子的胺类底物以及产物发生光催化氧化, 从而提高反应的产率. 其详细的反应机理如Scheme 18所示, 首先是激发态光催化剂可以与硫醚类催化剂PTH1发生单电子转移, 生成相应的硫自由基阳离子中间体和还原态光催化剂[IrII]. 还原态光催化剂可以将邻苯二甲酰亚胺衍生的活性酯18.1还原成相应的烷基自由基18.4. 烷基自由基18.4可以与硫自由基阳离子发生自由基-自由基偶联反应, 生成硫鎓离子中间体18.5. 该自由基偶联过程是经过DFT计算得以确认的, 其偶联过程的能垒更低. 硫鎓离子中间体18.5可以与芳基胺类底物18.2发生协同的SN2反应, 即可得到相应的C—N键偶联产物18.3. 反应过程中生成的硫鎓离子中间体则是通过高分辨质谱得以确定, 随后发生的亲核取代过程则是该反应的决速步.
2.2 光催化S—X键均裂产生硫自由基阳离子
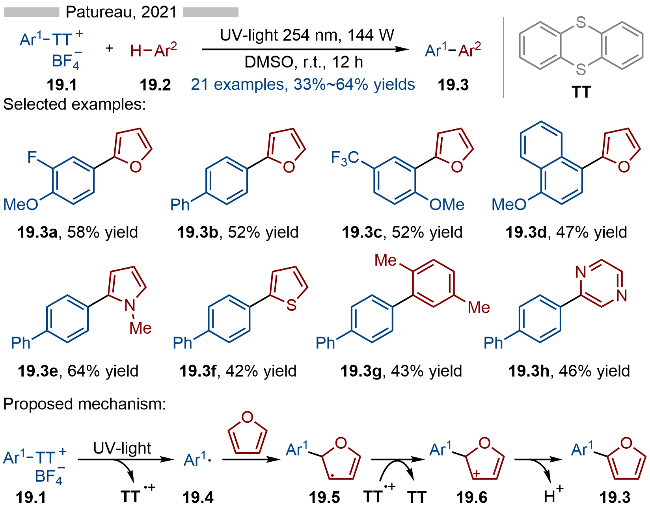
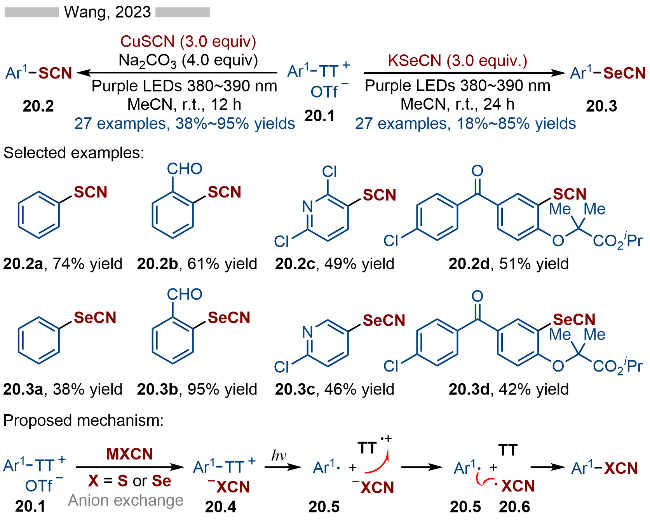
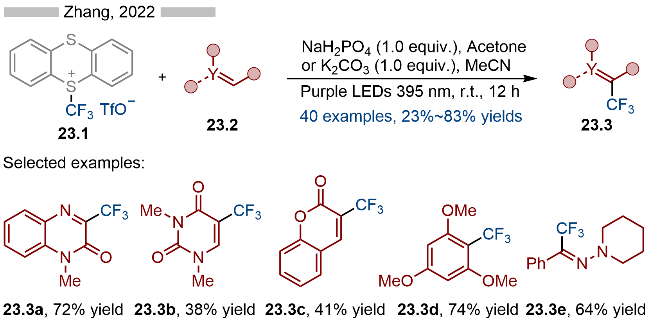
2023年, 王鹏课题组[32]报道了光催化芳基噻蒽盐类化合物的硫氰基化和硒氰基化反应, 并合成了一系列硫氰基和硒氰基取代的芳烃类化合物(Scheme 20). 在底物适用性方面, 无论是苯基取代还是氮杂芳烃取代的噻蒽类底物都能很好地适用于该反应. 此外, 药物分子—非诺贝特(Fenofibrate)衍生的噻蒽盐类底物也能很好地转化成相应的硫氰基和硒氰基取代的非诺贝特(产物20.2d和20.3d). 该策略在药物分子的后期修饰方面具有较高的应用前景. 其详细的反应机理如Scheme 20所示, 首先是噻蒽盐与NCX—发生阴离子交换, 生成相应的中间体20.4, 随后在光照条件下, 发生C—S键均裂, 生成相应的芳基自由基20.5和稳定的噻蒽自由基阳离子. 产生的噻蒽自由基阳离子可以将NCX—氧化成相应的自由基(NCX•), 再经过自由基-自由基偶联, 即可得到目标产物.
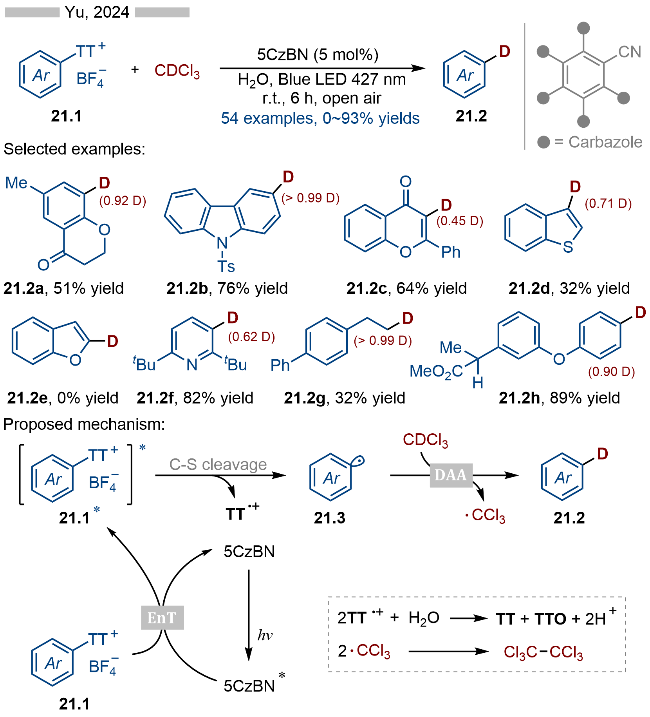
2024年, 於兵课题组[33]报道了能量转移催化噻蒽盐类化合物的氘化反应, 并合成了一系列氘代芳烃类化合物(Scheme 21). 该反应具有良好的底物适用性, 无论是苯基(产物21.2a)、咔唑(产物21.2b)、黄酮(产物21.2c)、苯并噻吩(产物21.2d)、吡啶(产物21.2f)甚至是烷基取代(产物21.2g)的噻蒽盐类底物都能适用于该反应. 不过富电子的杂芳烃类底物, 如呋喃和苯并呋喃衍生的噻蒽盐类底物则难以适用于该反应. 此外作者还对一系列含芳烃结构的药物分子进行了氘化反应, 以此合成一系列氘代的药物活性分子. 其详细反应机理如Scheme 21所示, 首先激发态的光催化剂5CzBN*与噻蒽盐类底物21.1发生能量转移, 从而生成相应的基态光催化剂5CzBN和激发态的噻蒽盐21.1*. 激发态的底物发生C—S键均裂, 生成相应的芳基自由基21.3和稳定的噻蒽自由基阳离子. 高活性的芳基自由基21.3可以与CDCl3发生氘原子转移反应(DAA), 生成相应的氘代芳烃类产物以及三氯甲基自由基. 反应体系中生成的噻蒽自由基阳离子则会被水淬灭掉, 而三氯甲基自由基则会发生二聚反应.
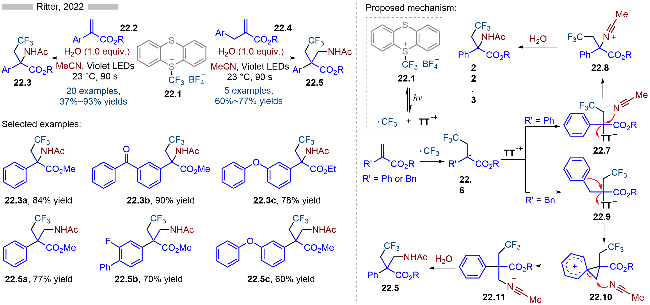
三氟甲基取代的噻蒽盐经过光催化同样也可以发生C—S键碎裂化, 并产生相应的三氟甲基自由基和噻蒽自由基阳离子. 基于该策略, Ritter课题组[34]报道了光催化丙烯酸酯类化合物的三氟甲基-氨基化反应, 并合成了一系列含季碳中性的α-和β-氨基酸酯类衍生物(Scheme 22). 当丙烯酸酯的α-位为芳基时, 则会生成相应的α-氨基酸酯; 当α-位为苄基结构时, 则会发生芳基迁移反应, 从而生成相应的β-氨基酸酯类化合物. 其详细的反应机理如Scheme 22所示, 首先光催化产生的三氟甲基自由基会与烯烃发生自由基加成反应, 生成羰基α-位烷基自由基22.6, 随后其与噻蒽自由基阳离子发生自由基-自由基偶联反应, 生成相应的噻蒽盐(22.7或22.9). 当取代基是苯基时, 则是中间体22.7. 该中间体在乙腈存在下, 发生亲核取代反应, 再经过水解等过程, 即可得到相应的α-氨基酸酯22.3. 当取代基为苄基时, 则会生成的相应的噻蒽离子中间体22.9. 在乙腈存在下, 中间体22.9发生分子内的芳基迁移反应, 再经过水解等过程, 即可得到相应的β-氨基酸酯22.5.
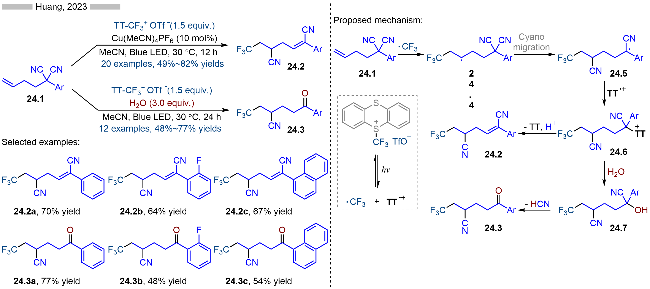
2023年, 黄良斌课题组[36]报道了光催化1,1-二腈类化合物的氰基迁移反应, 实现了烯烃的三氟甲基-氰基化(Scheme 24). 其中的三氟甲基试剂为相应的噻蒽盐类结构. 根据反应体系的不同, 则会生成不同的产物. 当反应体系中存在铜催化剂时, 则会生成(Z)-烯烃类产物24.2; 当反应中存在水分子时, 则会生成相应的酮类化合物24.3. 其详细的反应机理如Scheme 24所示, 首先三氟甲基取代的噻蒽盐类化合物经过光照后, 发生C—S键均裂, 生成相应的三氟甲基自由基和噻蒽自由基阳离子. 随后生成的三氟甲基自由基与烯烃反应, 生成烷基自由基24.4. 烷基自由基24.4经过五元环过渡态, 促使氰基发生迁移, 生成相应的苄基自由基24.5. 苄基自由基随后被稳定的噻蒽自由基阳离子捕获, 生成相应的噻蒽盐类中间体24.6. 反应条件不同, 产物也各不相同. 当发生消除反应时, 则会生成相应的烯烃类产物24.2; 当与水发生亲核取代反应时, 则会生成相应的醇24.7, 再经过β-位消除, 即可得到相应的酮类产物24.3.
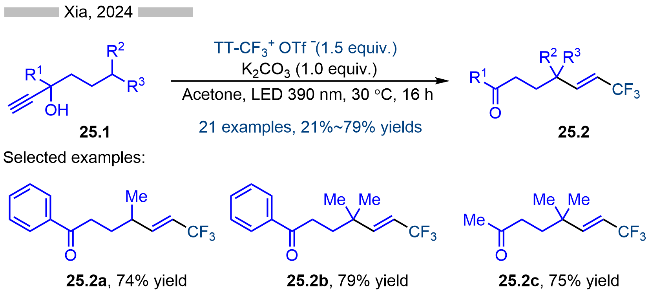
2020年, Ritter课题组[38]报道了光催化烯烃的氨基化反应, 并合成了一系列烷基取代的烯丙胺类化合物(Scheme 26). 该反应具有良好的底物适用性和官能团兼容性, 各种烷基取代的亚胺型噻蒽类底物、以及端烯和内烯自由基受体都能很好地适用于该反应. 其详细的反应机理如Scheme 26所示, 首先三氟乙酸酐在六氟异丙醇溶液中会分解产生大量的质子, 使得反应体系呈现酸性环境. 亚胺型噻蒽类底物26.2在酸性条件下发生质子化, 生成氨基取代的噻蒽阳离子26.4. 随后噻蒽阳离子26.4与激发态的光催化剂发生能量转移反应, 生成激发态的26.4*. 激发态的氨基噻蒽阳离子在酸性条件下发生N—S键均裂, 生成相应的氨基自由基阳离子26.5以及噻蒽自由基阳离子. 亲电性的氨基自由基阳离子则与烯烃发生加成反应, 生成氨基β-位烷基自由基26.6. 烷基自由基26.6与稳定的噻蒽自由基阳离子发生自由基-自由基偶联反应, 生成噻蒽盐类中间体26.7, 再经过消除反应/去质子化, 即可得到相应的烯丙胺类化合物26.3.
2.3 吩噻嗪类型光催化剂产生的硫自由基阳离子
吩噻嗪类化合物参与的光催化反应产生硫自由基阳离子的方式主要有两种, 其一是经过光照激发生成激发态的吩噻嗪, 再失去一个电子, 即可得到相应的硫自由基阳离子; 其二则是通过吩噻嗪类结构与缺电子底物形成电子给体-受体复合物(EDA复合物), 再经过光照发生单电子转移, 以此生成相应的硫自由基阳离子.
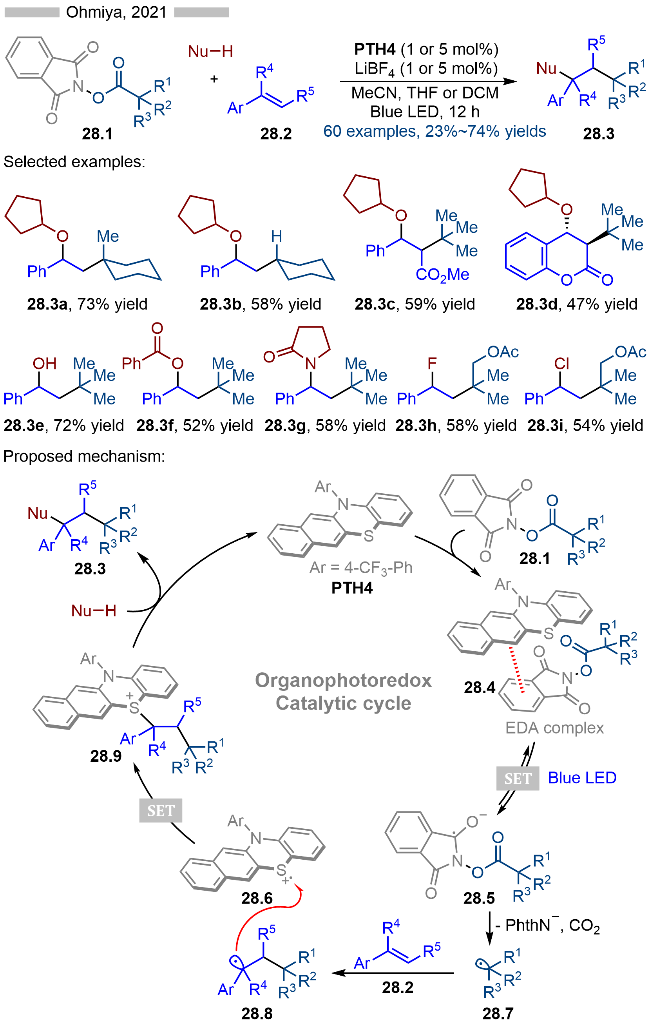
2020年, Ohmiya课题组[39]报道了基于N-芳基吩噻嗪类光催化剂催化的邻苯二甲酰亚胺衍生的羧酸酯类衍生物的脱羧偶联反应. 根据亲核试剂不同, 作者合成了一系列醚类、醇类、胺类以及硫醚类化合物(Scheme 27). 无论是三级烷基、二级烷基还是苄基羧酸衍生的活性酯都能很好地适用于该反应. 其详细的反应机理如Scheme 27所示, 首先基态光催化PTH在光照条件下, 生成激发态光催化剂. 激发态光催化剂与邻苯二甲酰亚胺衍生的羧酸酯类底物27.1发生单电子转移, 生成相应的烷基自由基27.4以及硫自由基阳离子PTH•+. 硫自由基阳离子与烷基自由基27.4发生单电子转移, 生成基态光催化剂PTH和碳正离子中间体, 随后碳正离子与PTH结合, 即可得到相应的硫鎓离子中间体27.5. 此外硫自由基阳离子PTH•+也可以与烷基自由基发生自由基-自由基偶联, 以此生成相应的中间体27.5. 最后亲核试剂27.2与硫鎓离子中间体27.5反应, 即可得到相应的目标产物27.3.
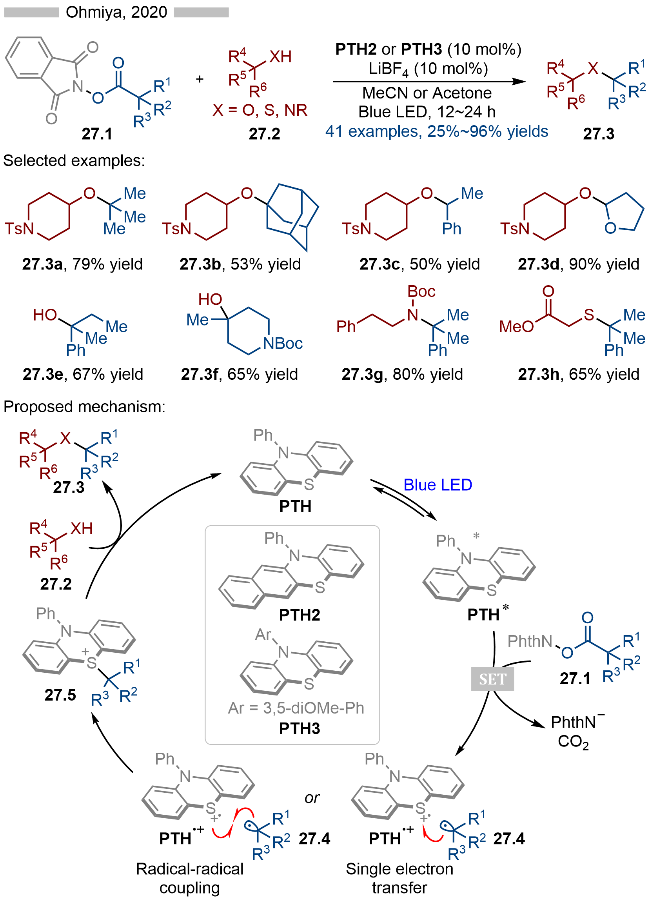
随后在2021年, Ohmiya课题组[40]将上述催化策略应用到烯烃的双官能团化反应中, 从而实现了C—C键和C—X键的构建(Scheme 28). 该反应同样以邻苯二甲酰亚胺衍生的羧酸酯类化合物为烷基自由基前体, 根据亲核试剂的不同, 作者分别实现了C—O键、C—N键、C—F键以及C—Cl键的构建. 在机理研究方面, 作者通过紫外可见吸收光谱, 证明了PTH4可以与缺电子的活性酯28.1形成EDA复合物. 在光照条件下EDA复合物发生单电子转移, 生成相应的邻苯二甲酰亚胺自由基阴离子28.5和硫自由基阳离子28.6. 中间体28.5发生碎裂化, 生成烷基自由基28.7, 其与苯乙烯类型底物28.2发生自由基加成反应, 生成相应的苄基自由基28.8. 在该反应中, 作者认为苄基自由基会被硫自由基阳离子氧化成相应的碳正离子中间体, 再经过C—S键偶联, 得到相应的硫鎓离子中间体28.9. 硫鎓离子中间体则是通过高分辨质谱得以确认. 最后中间体28.9与亲核试剂发生反应, 即可得到相应的目标化合物28.3.
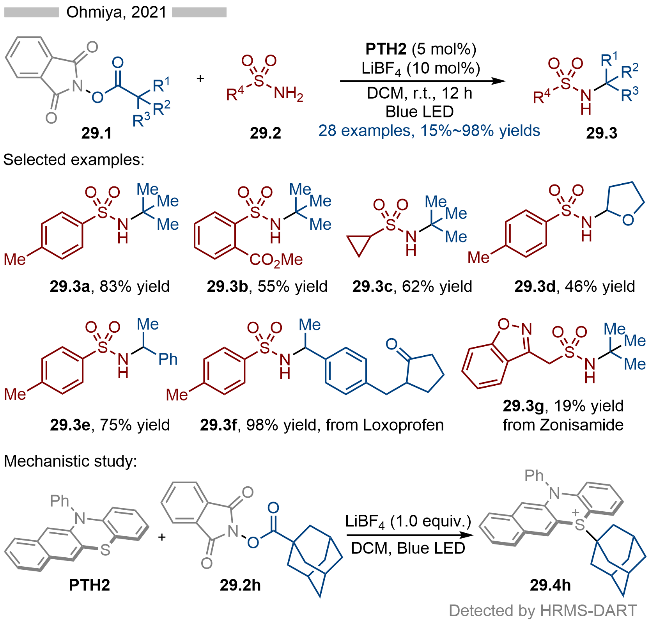
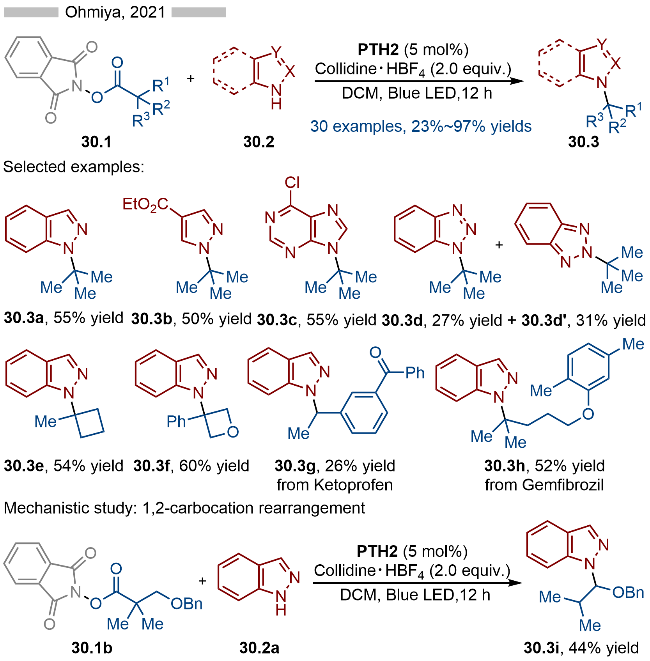
同年, Ohmiya课题组[42]基于相同的光催化反应策略, 实现了唑类芳烃的N-烷基化(Scheme 30). 该反应同样以邻苯二甲酰亚胺衍生的羧酸酯为烷基化试剂, 通过其与催化剂PTH2形成EDA复合物. 随后在光催化条件下EDA复合物发生碎裂化, 生成烷基自由基. 烷基自由基再通过单电子氧化/C—S形成等过程, 即可得到相应的烷基取代的硫鎓离子中间体. 最后通过亲核取代反应, 生成相应的目标产物. 反应具有良好的底物适用性, 无论是吲唑(产物30.3a)、吡唑(产物30.3b)还是嘌呤(产物30.3c)都能很好地实现其N-烷基化反应. 当为苯并三氮唑底物时, 则会存在一定的区域选择性(产物30.3d和30.3d'). 此外药物分子衍生的活性酯(产物30.3g和30.3h)也能很好地实现其脱酸氨基化反应. 在机理研究方面, 作者通过1,2-碳正离子重排反应, 表明该过程存在碳正离子或碳正离子等价物这一中间体.
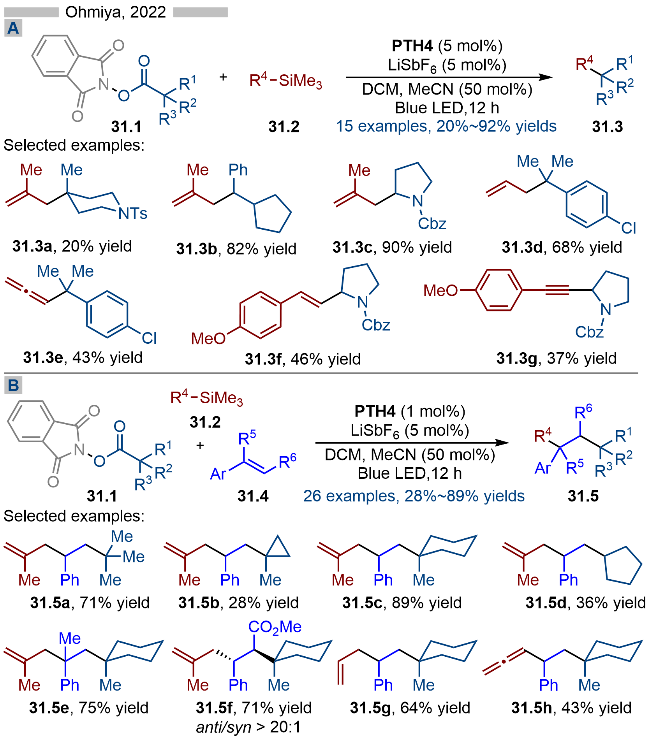
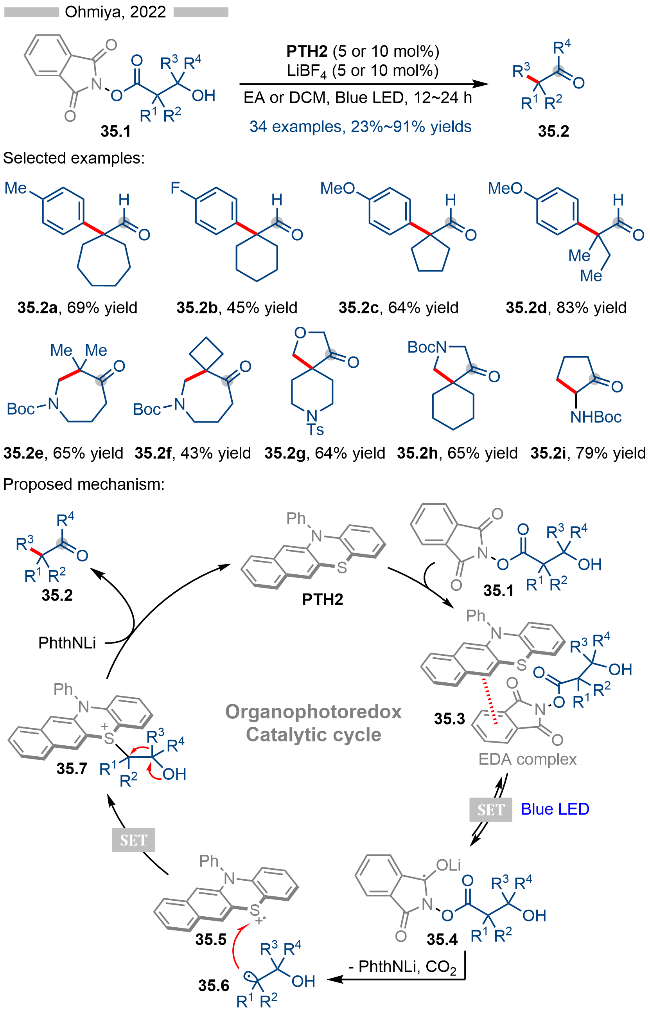
2022年, Ohmiya课题组[43]又报道了基于光催化羧酸类衍生物的脱酸官能团化反应(Scheme 31A). 基于不同的亲核试剂, 作者实现了脱羧烯丙基化(产物31.3a~31.3d)、联烯基化(产物31.3e)、烯基化(产物31.3f)以及炔基化(产物31.3g)反应. 随后作者将该策略应用到三组分串联反应中, 以此实现了烯烃的双官能团化 (Scheme 31B). 其反应机理与Scheme 28中类似, 其亲核试剂则是相应的三甲基硅基衍生的化合物.
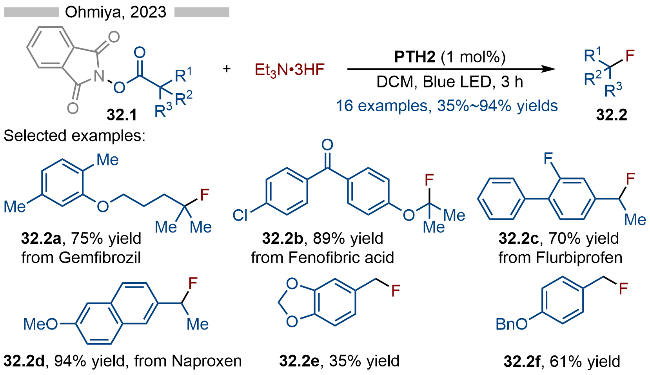
核苷酸类药物分子经过化学修饰以后能够有效增强其代谢稳定性以及药效活性, 因此对于该方向的研究一直备受关注. 其中烷基膦酸酯类结构是亚膦酸酯类结构理想的中性替代物. 目前引入的烷基部分主要是甲基、一级烷基或二级烷基结构, 而对于大位阻的三级烷基膦酸酯衍生的核苷酸类化合物的合成相对较少. 2023年, Ohmiya课题组[45]基于前期的研究基础, 通过光催化实现了含核苷酸的亚膦酸酯类结构的三级烷基化反应, 并合成了一系列烷基膦酸酯衍生的核苷酸类化合物(Scheme 33). 该反应以邻苯二甲酰亚胺衍生的羧酸酯类化合物为相应的烷基化试剂, 以2-氰基-1-苯乙基为相应的离去基团, 经过光催化, 实现C—P键的构建. 反应具有极高的官能团兼容性和底物适用性. 其详细的反应机理如Scheme 33所示, PTH5与活性酯形成EDA复合物, 再经过光照, 发生单电子转移, 以此生成相应的中间体33.5和硫自由基阳离子33.6. 中间体33.5经过碎裂化, 生成相应的烷基自由基33.7, 再经过单电子氧化/C—S键形成, 即可得到相应的硫鎓离子中间体33.8. 亚膦酸酯类底物与中间体33.8发生Michaelis-Arbuzov类型反应, 即可得到相应的烷基膦酸酯类产物33.3.
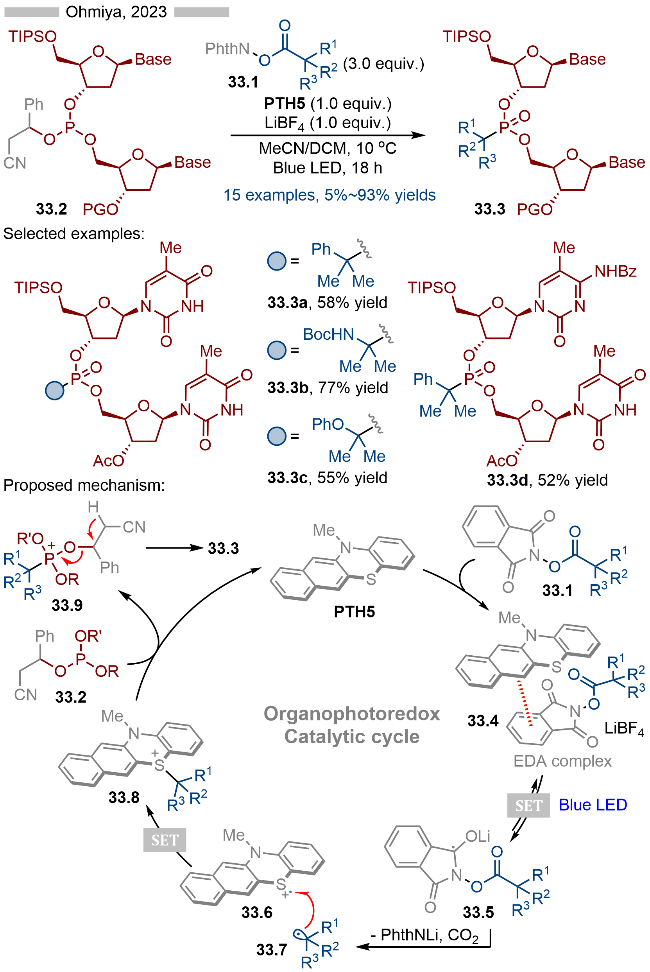
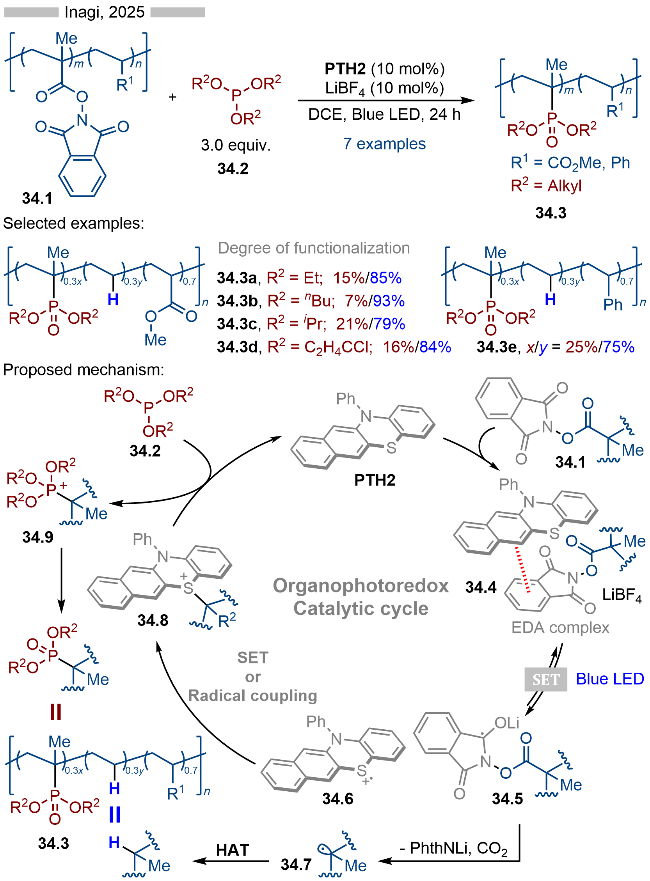
2025年, Inagi课题组[46]报道了基于光催化聚丙烯酸酯类衍生物的脱羧官能团化反应, 并合成了一系列含膦酸酯结构的聚合物(Scheme 34). 该策略既保持了原有的聚合度, 又重新构建了新的多聚物. 在反应过程中, 其结构中不可避免地存在这一定量的脱羧氢化反应. 其详细的反应机理如Scheme 34所示, 首先PTH2与聚合物分子上的活性酯部分形成EDA复合物, 随后在光照条件下, 发生单电子转移, 生成相应的中间体34.5和硫自由基阳离子34.6. 中间体34.5发生碎裂化, 产生烷基自由基34.7, 烷基自由基可以直接与聚合物发生氢原子转移, 以此实现其氢化过程. 此外生成的烷基自由基34.7还可以与硫自由基阳离子发生单电子转移/C—S键偶联, 生成相应的硫鎓离子中间体34.8. 中间体34.8也可能是通过自由基-自由基偶联的方式产生的. 生成的硫鎓离子中间体34.8等效于碳正离子中间体, 其可以与亚磷酸酯反应, 生成相应的中间体34.9, 再经过后续转化, 即可得到相应的含膦酸酯结构的聚合物34.3.
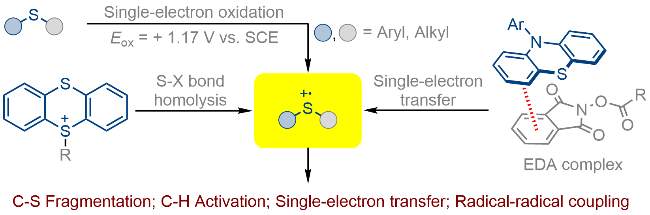
硫自由基阳离子可以参与一系列光催化的单电子氧化以及自由基-自由基偶联, 以此实现碳正离子中间体或硫鎓离子中间体的合成, 从而为后续反应提供活性位点.
3 总结与展望
总的来说, 硫自由基阳离子的产生方式主要包括以下三种途径(Scheme 36): 其一是硫醚类化合物的光催化单电子氧化, 其产生的硫自由基阳离子可以实现C—S键碎裂化以及C—H键活化; 其二是噻蒽盐类化合物的S—X键均裂, 其产生的噻蒽自由基阳离子可以参与后续的单电子转移、羟基活化以及自由基-自由基偶联等过程; 其三则是吩噻嗪类光催化剂的光化学氧化, 其产生的硫自由基阳离子可以参与后续的单电子氧化以及自由基-自由基偶联等过程. 尽管硫自由基阳离子的产生方式相对有限, 但其展现出来的反应模式却多种多样: 其一是通过C—S键断裂化, 生成亲电性阳离子中间体, 以此实现其官能团化反应; 其二则是与氮自由基阳离子类似, 可选择性地实现其α-位C—H的官能团化; 其三则是与膦自由基阳离子类似, 可以与羟基结合, 以此实现醇类底物的活化; 其四则是与中性自由基类似, 实现自由基-自由基偶联反应; 最后硫自由基阳离子可以作为单电子氧化剂, 参与到光化学反应中.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
尽管硫自由基阳离子领域已取得了突破性进展, 但其反应机制与应用仍存在诸多关键科学问题亟待突破: 其一是中间体形成机制不明晰, 当前对硫鎓离子中间体的生成过程缺乏系统性研究, 反应机理在单电子转移/C—S键偶联模式与自由基-自由基偶联模式之间存在争议. 其二是反应的选择性问题缺乏系统性研究, 硫自由基阳离子虽能能够参与多种反应模式, 但选择性调控机制尚未阐明. 其三是应用潜力亟待开发, 其在不对称催化领域的研究以及电化学领域有着较强的应用前景, 此外在多肽类化合物的选择性修饰方面具有潜在的应用价值.
(Cheng, B.)