

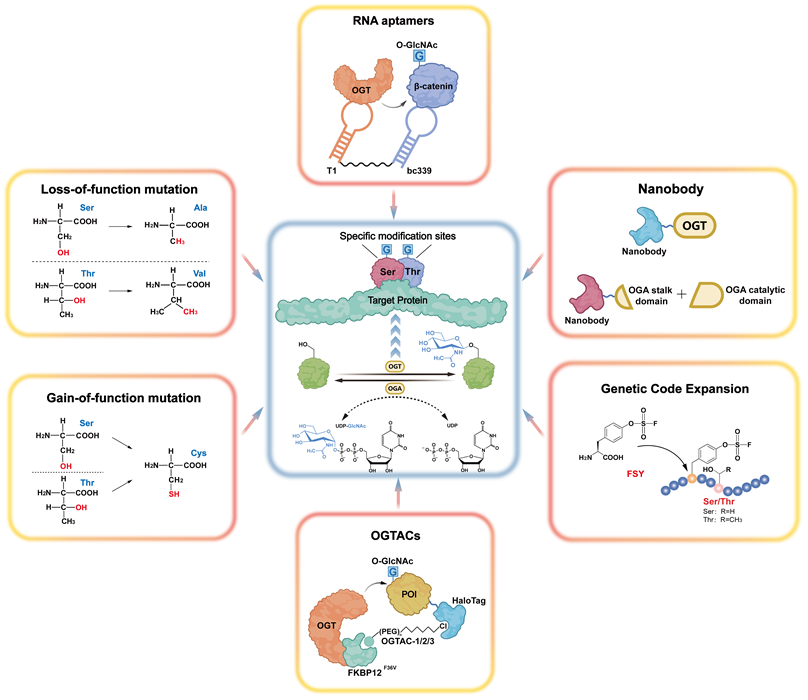
1 引言
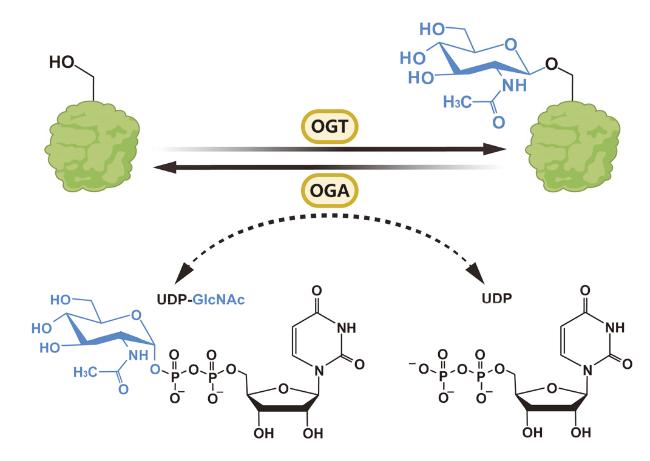
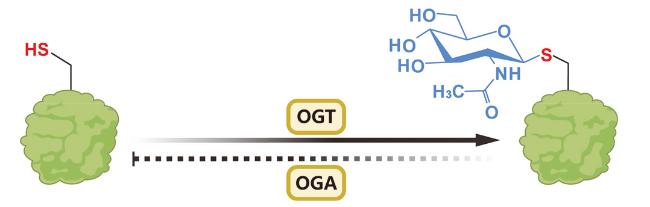
蛋白质O-连接β-N-乙酰葡萄糖胺(O-GlcNAc)糖基化是一种高度动态可逆的单糖修饰[1], 是十分重要的蛋白质翻译后修饰之一. 已报道的O-GlcNAc糖基化蛋白超过5000种, 广泛分布于真核细胞的细胞质[2]、细胞 核[2]、线粒体[3]和区室质膜[4]上. O-GlcNAc糖基化以己糖胺生物合成途径(HBP)终产物UDP-GlcNAc为糖供体, 由糖基转移酶OGT负责将GlcNAc转移到底物蛋白的丝氨酸(Ser)残基和苏氨酸(Thr)残基的羟基氧上, 由糖基水解酶OGA负责将GlcNAc水解下来[5-6](图1). O-GlcNAc糖基化参与调控基因转录、表观遗传修饰等基本细胞过程及多种信号事件[7], 异常的蛋白质O-GlcNAc糖基化水平是人类疾病发生发展的关键, 包括糖尿病[8]、癌症[9-11]和神经退行性疾病[12-14]等.
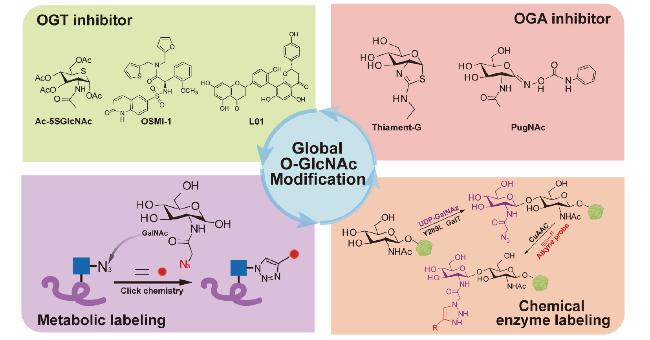
近年来, 关于O-GlcNAc修饰的研究方法的开发取得了飞速进展, 为我们理解O-GlcNAc修饰在细胞中的 作用提供了重要工具(图2). 化学小分子抑制剂作为其中的一种关键工具, 广泛应用于探索O-GlcNAc修饰的功能与机制. 例如, OGT抑制剂Ac-5SGlcNAc、OSMI-1、L01通过抑制OGT酶活性降低O-GlcNAc修饰水平, 影响肿瘤细胞代谢; OGA抑制剂Thiamet-G、PugNAc通过抑制OGA酶活性增强O-GlcNAc修饰水平, 应用于阿尔茨海默病等神经退行性疾病及肿瘤的研究[15]. 此外还有代谢标记技术, 特别是Boyce等[16]提出的非天然糖供体标记法, 通过引入叠氮标记的GlcNAz并结合生物正交反应, 能够高效标记O-GlcNAc修饰的蛋白质, 避免了传统方法中的非特异性干扰. 除了代谢标记法, 化学酶标记法也在O-GlcNAc修饰的研究中发挥了重要作用. 化学酶标记法通过糖基转移酶将带有生物正交基团的非天然糖探针引入O-GlcNAc修饰的底物蛋白中. 该方法的优势在于能够在体外进行, 避免了非天然糖探针对细胞生理活动的干扰. 通过这种方法, 研究者能够对O-GlcNAc修饰的蛋白进行捕获、富集和检测. Khidekel 等[17]利用工程化β-1,4-半乳糖转移酶GalT1 (Y289L)扩展的底物耐受性, 将携带酮基化学手柄的半乳糖类似物选择性链接至O-GlcNAc修饰的蛋白质上. 随后, 引入的酮基通过与氨基氧生物素发生肟化反应, 实现对O-GlcNAc修饰蛋白的生物素化标记. 最终, 利用链霉亲和素-HRP结合化学发光检测, 实现了对O-GlcNAc修饰蛋白的高灵敏、高特异性检测. Griffin等[18]将叠氮修饰的UDP-N-叠氮乙酰半乳糖胺(UDP-GalNAz)作为糖供体, 通过Gal-T1(Y289L)催化可以高效标记O-GlcNAc修饰的蛋白并进行后续分析. 在Wen等[19]的研究中, β-(1,4)-N-乙酰半乳糖胺转移酶(CgtA)的催化下, UDP-GalNAz还可通过两步反应实现特异性检测N-乙酰神经氨酸-α(2-3)-半乳糖(Neu5Acα(2-3)Gal)糖链结构. 除了UDP-GalNAc, Li等[20]开发了新型代谢标记探针Ac34dGlcNAz, 其通过C4位脱氧设计有效避免非特异性糖缀合物标记, 并利用其抗OGA水解的特性显著提升O-GlcNAc修饰蛋白的标记效率与稳定性, 在细胞成像和蛋白质组学分析中展现出高选择性和高效能, 为O-GlcNAc研究提供了更可靠的工具. 尽管这些方法在调控O-GlcNAc修饰方面取得了重要突破, 但由于O-GlcNAc修饰的动态性和复杂性, 这些方法可能会同时改变细胞内数千种蛋白质的O-GlcNAc修饰水平, 这使得研究O-GlcNAc糖基化修饰如何精准调控特定蛋白上特定修饰位点的生物学功能面临挑战. 因此, 在不影响细胞内全局糖基化水平的情况下, 开发靶向改变特定蛋白质的O-GlcNAc修饰的工具和方法是十分必要的. 本文主要总结了近年来在靶向蛋白质O-GlcNAc糖基化领域中不断发展的研究工具与技术手段, 为探索O-GlcNAc修饰的精确调控功能提供了可能. 另外, 基于现有技术方法, 并结合最新的研究进展, 本文探讨了O-GlcNAc修饰位点在疾病调控中的功能意义, 旨在为后续相关研究提供参考和启发.
2 靶向特定蛋白质的O-GlcNAc糖基化精准调控工具
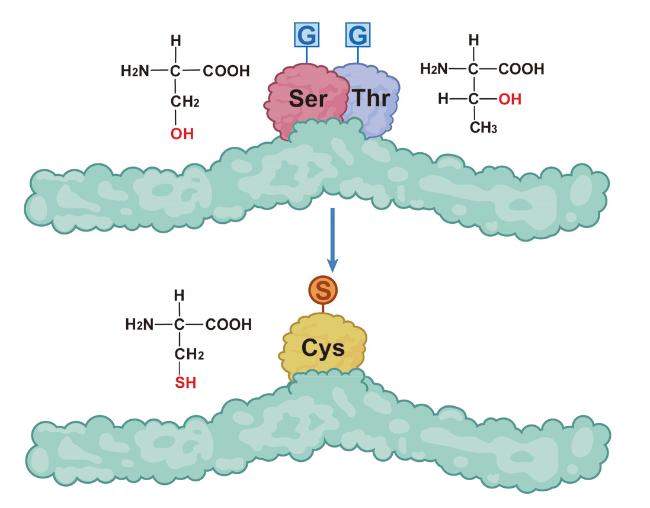
2.1 功能丧失性O-GlcNAc修饰位点突变
在多样的蛋白质翻译后修饰中, O-GlcNAc糖基化与磷酸化这两种动态可逆的修饰方式在Ser/Thr位点上存在竞争性修饰现象, 已成为蛋白功能研究的重要挑 战[21-22]. 研究者开发了“磷酸化模拟突变”策略, 通过将Ser/Thr替换为带负电荷的天冬氨酸(Asp), 利用电荷相似性模拟磷酸基团引入的静电效应[23]. 这种突变体不仅能稳定保持类磷酸化状态, 规避体内磷酸化/去磷酸化动态循环的干扰, 还能有效模拟磷酸化对蛋白质构象、亚细胞定位及相互作用网络的调控作用. “磷酸化模拟突变”策略已被用于研究蛋白质磷酸化, 但尚无模拟氨基酸能够准确地直接代表O-GlcNAc修饰的氨基酸残基. 目前, 功能丧失性突变被用于研究位点特异性O-GlcNAc修饰的缺失对蛋白质功能的影响. O-GlcNAc修饰发生在蛋白质Ser和Thr残基的羟基氧上, 当使用氢原子取代Ser羟基侧链, 使Ser突变为丙氨酸(Ala), 或使用甲基基团取代Thr羟基侧链, 使Thr突变为缬氨酸(Val), 即可将O-GlcNAc修饰位点突变(图3). 这两种突变方式均可阻断内源性O-GlcNAc修饰, 达到“敲除”蛋白质特定位点O-GlcNAc修饰的效果.
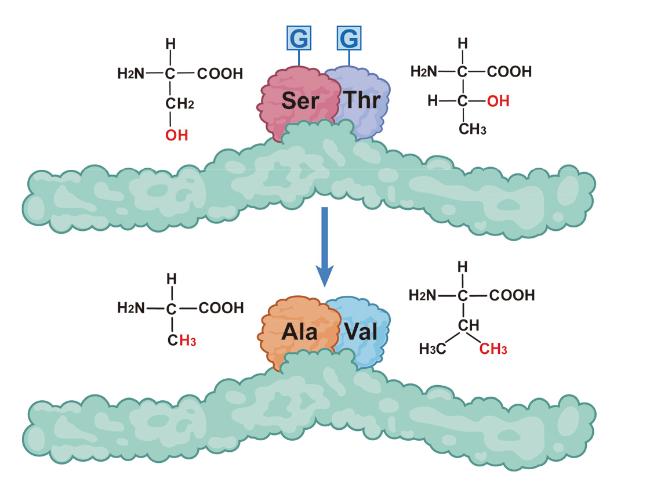
这种突变策略可以与质谱联用来确定蛋白质的O-GlcNAc修饰位点. Liu等[24]在探索O-GlcNAc修饰调控先锋转录因子FOXA1功能的研究中, 通过该策略确定了FOXA1的三个糖基化修饰位点. 该研究通过质谱分析发现了FOXA1的三个O-GlcNAc修饰位点, 之后构建了位点突变质粒, 将这三个位点的氨基酸突变为Ala, 并在细胞内进行异位表达. Western Blotting结果证明, 位点突变FOXA1的O-GlcNAc修饰水平明显低于野生型, 三位点突变FOXA1的O-GlcNAc修饰几乎全部消失, 证实这三个位点均为FOXA1的O-GlcNAc修饰位点. Ogawa等[25]在体外研究了O-GlcNAc修饰对折叠表皮生长因子样结构域稳定性和Notch1转运的影响, 将表皮生长因子EGF结构域26-33 位Thr突变为Val, 导致细胞表面FLAG-Notch1119-1916的表达下降. 此外, 该技术还可用于比较同一蛋白的不同O-GlcNAc修饰位点对其功能的影响. 一项针对突变SMAD4的四个O-GlcNAc修饰位点的研究表明, Thr63 位的O-GlcNAc修饰对维持SMAD4稳定最为重要[26]. Ma等[27]的研究表明, 在NF-κB上丢失一个高度保守的O-GlcNAc修饰位点(T305)会通过升高另一个位点(T352)的O-GlcNAc修饰水平来进行补偿. 这种位点特异性的差异凸显了对O-GlcNAc修饰进行分子水平研究的价值. 此外, 质谱分析作为这种突变策略的互补技术, 能够揭示在之前研究中可能被忽视的修饰差异.
然而, 这一策略无法直接靶向改变细胞内源蛋白的O-GlcNAc修饰, 只能通过体外构建突变质粒再进行异位表达来改变蛋白的糖基化水平. 另外需要注意的是, 在这种突变策略中, 将极性氨基酸替换为非极性氨基酸, 可能会对蛋白质的结构和功能产生一些影响. 同时, 考虑到磷酸化和O-GlcNAc修饰之间的串扰, Ser/Thr突变为Ala/Val也会对蛋白质的磷酸化产生影响.
2.2 功能获得性O-GlcNAc修饰位点突变
De Leon等[29]通过体外合成α-突触核蛋白的S-GlcNAc修饰, 证明了该修饰能够抵抗OGA的水解, 其对蛋白质的修饰效应与同一位点的O-GlcNAc修饰相似. 此外, Tegl等[30]设计了一种硫醇糖基化酶, 通过突变OGT的催化活性残基, 能够将GlcNAc基团特异性地连接到Cys上. 该酶促反应使用商用糖基供体pNP-GlcNAc, 显著提高了技术的可操作性和便利性. 研究人员通过S-GlcNAc化的tau蛋白作为验证, 进一步展示了该技术的有效性. Gorelik等[31]开发了一种技术, 通过CRISPR-Cas9技术在哺乳动物系统中将目标蛋白的Ser/Thr突变为Cys, 从而改变特定蛋白的O-GlcNAc修饰, 而不影响全局的O-GlcNAc糖基化水平. S-GlcNAc修饰可通过具备某些位点特异性和广泛选择性的O-GlcNAc抗体(如CTD110.6)进行检测. 该团队首先在体外验证了OGT对多种底物序列均具有S-GlcNAc转移酶活性, 随后将该方法应用于哺乳动物细胞中. 他们利用CRISPR-Cas9技术将小鼠胚胎干细胞中OGA上O-GlcNAc修饰位点(S405C)突变. 由于缺乏针对O-GlcNAc修饰OGA的特异性抗体, 他们通过在细胞裂解液中进行GalTY289L标记, 随后使用GalNAz进行点击化学反应, 并与炔基标记的PEG 5000结合, 量化OGA上GlcNAc修饰的化学计量比. 研究结果显示, S-GlcNAc的掺入率至少达到了70%. 这种方法有望通过人为增加特定位点的修饰比例, 来深入研究活体系统中蛋白质的O-GlcNAc修饰. 然而, 在荧光偏振竞争测定中, S-GlcNAc修饰肽段与OGA的结合亲和力相比 O-GlcNAc修饰肽段下降了两个数量级. 这种识别能力的下降表明, S-to-C的突变可能影响其他相互作用分子对该修饰的识别, 这在使用该技术进行研究时可能会带来复杂性. 最近, Mitchell等[32]又开发了一种高效生成均一的类O-GlcNAc修饰蛋白新技术, 专用于体外研究. 基于他们之前的研究, 他们利用OGT对Cys替代Ser的高耐受性, 结合共表达OGA, 实现了位点特异性且高度均一的S-GlcNAc单糖基化. 该技术应用于DDX3X、TAB1和CK2α, 研究结果表明, 这些蛋白质的近乎均一的单S-GlcNAc修饰显著提升了DDX3X和CK2α的溶解性. 此外, 他们还成功获得了单S-GlcNAc修饰的TAB1晶体, 尽管其衍射质量有限.
虽然Ser和Cys两者结构相似, 但在这种功能获得性突变策略中也可能带来不可忽视的功能性影响. 一方面, 这种突变策略不仅阻断磷酸化修饰与O-GlcNAc修饰之间的串扰调控, 还会因引入巯基诱发形成二硫键或蛋白质错误折叠[33]. 另一方面, S-GlcNAc修饰不可被OGA水解, 从而形成一种持久性修饰状态. 因此, S-to-C突变并不等同于简单的“去除O-GlcNAc”, 而是用一种不可逆糖基化修饰取代了可逆修饰, 这可能导致蛋白功能被持续激活或抑制, 甚至产生新的调控机制.
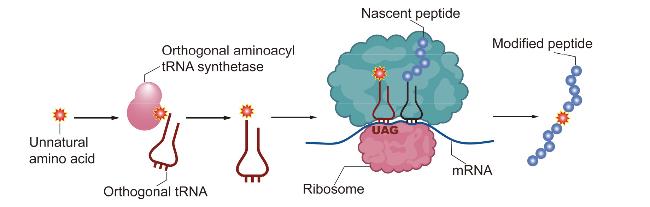
2.3 遗传密码扩展引入位点特异性O-GlcNAc修饰
遗传密码扩展(Genetic Code Expansion, GCE)是一项强大的生物技术, 它通过在目标基因的指定位置引入琥珀型终止密码子(UAG), 利用正交氨酰化-tRNA合成酶和正交tRNA(这种合成酶不会对细胞内正常的tRNA进行氨酰化, 同时正交tRNA也不是细胞中正常合成酶的底物), 在翻译过程中特异性地将非天然氨基酸(UAAs)引入蛋白质中, 实现对蛋白质功能的精细调控和定点修饰[34](图6). 该技术能够以定量且特异的方式在细胞内表达蛋白质, 而不干扰整体蛋白组. 由此, 一种可能的间接策略是通过GCE技术首先向蛋白中引入反应性基团(如炔基[35]或酮基[36])的UAAs, 然后通过点击化学反应与带有叠氮基团的GlcNAz偶联, 以实现在蛋白质特定位点引入O-GlcNAc修饰, 然而这种方法所产生的非天然糖苷键是否能够代表生理状态下天然形式的糖基化, 是一个值得探究的问题.
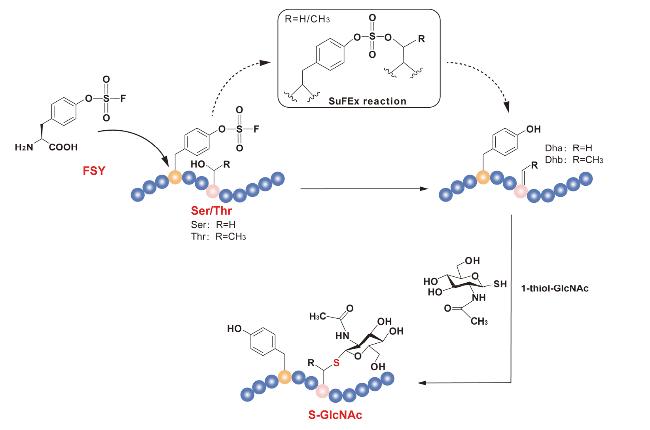
由于翻译机制的固有限制以及对生物环境兼容性的要求, 通过UAAs引入的功能性基团目前仅限于化学惰性、生物正交或潜在的生化活性基团. 为了突破这一障碍, Yang等[37]结合GCE和化学转化提出了GECCO (Genetically Encoded Chemical COnversion)创新策略, 该策略结合了GCE技术和硫氟交换(SuFEx)反应, 通过在活细胞中将具有生物反应性的UAAs(如氟磺酸酪氨酸FSY)引入到靠近目标天然氨基酸(如Ser和Thr)的位置, 使两者发生SuFEx反应, 生成反应性脱氢丙氨酸(Dha)或脱氢丁氨酸(Dhb), 进而在体外与硫醇化糖类(1-thiol-GlcNAc)反应形成稳定的S-GlcNAc修饰(图7). 这种方法旨在在体外模拟O-GlcNAc的功能, 但当前应用仅限于体外纯化的蛋白质, 尚未完全实现在细胞内的应用.
迄今为止, 尚无研究成功通过GCE技术将合成的O-GlcNAc修饰氨基酸引入蛋白质中. 最早一项研究曾报道利用UAG抑制系统将O-GlcNAc-Thr引入到肌红蛋白中, 但该研究后被撤回. 此外, 将GCE应用于O-GlcNAc研究的一大障碍在于对合成O-GlcNAc修饰氨基酸的摄取与稳定性. 存在O-GlcNAc修饰的Ser会被大肠杆菌(E. coli)代谢为碳源, 因此在使用UAG抑制系统时, 其无法在细胞质中作为稳定的UAAs[38-39]. 为提高细胞摄取效率而设计的全乙酰化的O-GlcNAc修饰Ser变体, 在大肠杆菌中无法被去乙酰化, 因为大肠杆菌不表达所需的脱乙酰酶[40]. 相比之下, 这种策略更适用于具备广谱脱酰化酶系统的哺乳动物细胞.
2.4 基于纳米抗体的O-GlcNAc修饰靶向调控
Ramirez等[44]开发了基于纳米抗体的O-GlcNAc修饰靶向调控系统, 实现了对特定蛋白O-GlcNAc修饰的精准编辑, 为O-GlcNAc修饰功能的研究提供了新范式. 该系统通过将OGT与特异性识别靶标标签(如GFP或四肽EPEA)的纳米抗体融合, 构建“邻近导向型OGT”, 利用纳米抗体的靶向性, 将OGT定位至目标蛋白(图8a). nGFP-OGT融合体可特异性修饰GFP标记的JunB、cJun等转录因子, nEPEA-OGT融合体则靶向修饰EPEA标签融合的核孔蛋白Nup62. 但实验结果显示nGFP-OGT融合体靶向修饰目标蛋白的同时, 也扰动了全局O-GlcNAc修饰水平. 为提升nGFP-OGT融合体的选择性, Ramirez等[44]截短了OGT的TPR结构域, 仅保留前4个重复单元, 显著提升了其靶向性, 同时通过定量糖蛋白质组学证实了其靶标修饰的特异性. 该系统成功实现了靶向催化内源性α-突触核蛋白发生O-GlcNAc修饰, 为神经退行性疾病研究提供了新工具. 另外, 为靶向去除O-GlcNAc修饰, Ge等[45]又研发了Nanobody- OGA融合体. 通过优化OGA结构, 将其催化结构域和茎结构域分离截短, 并将茎结构域与纳米抗体融合, 形成nGFP-splitOGA融合体(图8b). 实验结果显示, 在纳米抗体引导下, splitOGA的催化结构域和茎结构域片段在目标蛋白附近重组, 恢复OGA活性, 可特异性去除GFP-Nup62、GFP-Sp1等靶蛋白的O-GlcNAc修饰, 而对CREB等内源性蛋白的修饰无显著影响. 进一步通过14肽Ubc标签系统验证了该策略的通用性, 并发现O-GlcNAc修饰缺失会加速c-Jun降解.
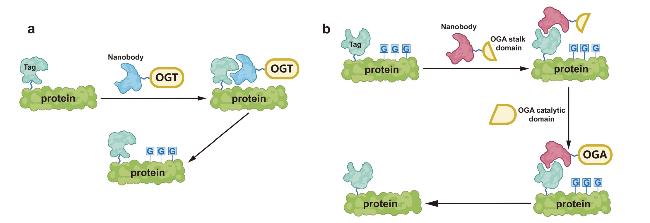
图8 基于纳米抗体的O-GlcNAc修饰靶向调控: (a) OGT通过标签纳米抗体空间接近靶蛋白, 使融合表达标签的靶蛋白O-GlcNAc修饰增加. (b) OGA通过标签纳米抗体去除靶蛋白O-GlcNAc修饰. OGA茎和催化结构域的分裂可抑制脱靶活性Figure 8 Nanobody-based targeted regulation of O-GlcNAcylation. (a) OGT is recruited into proximity of the target protein via a tag-specific nanobody, leading to increased O-GlcNAcylation on the tagged target protein. (b) OGA removes O-GlcNAc from the target protein via the tag-specific nanobody. Splitting the OGA stalk and catalytic domains can suppress off-target activity |
这两套纳米抗体系统均展现出高度的灵活性和兼容性, 支持GFP、EPEA、Ubc、BC2等多种标签, 通过更换纳米抗体可以适配不同靶标蛋白. 其次, 该系统通过引入催化失活突变体(如OGT-K852A、OGA-D174N)作为阴性对照, 排除了纳米抗体本身对靶标功能的干扰. 结合化学酶标记与串联质谱标签(TMT)定量技术, 该系统也实现了对单蛋白修饰位点分析和全局糖蛋白质组监测. 这些工具突破了传统突变策略的局限, 尤其适用于对多修饰位点蛋白或未确定修饰位点蛋白的功能研究. 未来可开发内源性蛋白靶向纳米抗体, 减少对外源标签依赖, 或结合化学/光遗传学控制纳米抗体活性, 实现时空特异性调控, 甚至拓展至其他糖基转移酶, 构建更广泛的翻译后修饰编辑平台.
2.5 靶向特定蛋白O-GlcNAc修饰的嵌合体分子
受化学诱导的蛋白质翻译后修饰方法的启发, Ma等[46]首次提出OGTACs (O-GlcNAcylation Targeting Chimeras), 一种能够将OGT招募到特定蛋白上的异双功能小分子系统. OGTACs基于化学诱导邻近(Chemically Induced Proximity, CIP)策略, 通过桥接OGT与靶蛋白, 实现靶蛋白位点特异性O-GlcNAc修饰, 为蛋白质O-GlcNAc修饰的功能研究和疾病治疗提供了新工具. OGTACs在结构上可分为OGT招募域和靶蛋白结合域. 其中OGT招募域由小分子配体AP1867组成, 可与FKBP12F36V融合表达的OGT(fOGT)特异性结合. 靶蛋白结合域由卤代烷基团组成, 能够共价结合HaloTag融合表达的靶蛋白(如BRD4、CK2α). OGT招募域和靶蛋白结合域通过不同长度的聚乙二醇(PEG)连接子连接, 形成OGTAC-1/2/3一系列分子(图9a). 通过优化PEG单元数调控OGT与靶蛋白的空间距离, 可以显著影响OGT催化效率, 短链接的OGTAC-1催化效率显著优于长链接的OGTAC-2/3. 另外OGTAC-4与OGTAC-1/2/3稍有不同, 在结构上, OGTAC-4的靶蛋白结合域由JQ1配体组成, 以非共价结合的方式靶向内源性蛋白质BRD4的BD1/BD2结构域, 实现内源蛋白修饰. 受先前研究的启发, Ma团队[46]又开发了截短型fOGT(tfOGT). 其中, 0tfOGT是经筛选得到的最优构建体, 在高表达水平下, 其显示对总体O-GlcNAc修饰水平干扰最小. 接下来将OGTAC-4应用于0tfOGT:HTN-BRD4系统, 结果显示BRD4的O-GlcNAc修饰水平增加5倍, 证实了OGTAC-4能够有效利用0tfOGT的活性来诱导BRD4发生O-GlcNAc修饰, 而不破坏总体O-GlcNAc修饰水平(图9b). OGTACs具有良好的靶向效能, LC-MS/MS鉴定显示, OGTACs可诱导BRD4在C端区域(S1051 to F1224)发生特异性O-GlcNAc修饰, OGTAC-1可选择性修饰HTN-CK2α的S347位点且几乎不扰动内源性CK2α的O-GlcNAc修饰水平, OGTACs也成功应用于EZH2等蛋白. 目前已有超过20,000种HaloTag 融合的人类蛋白质质粒, OGTACs可广泛应用于特定蛋白的O-GlcNAc修饰, 具有广谱适用性. OGTACs还可通过调整浓度和时间参数, 实现动态调控靶蛋白的O-GlcNAc修饰水平. 通过AP1867竞争性结合, 可6 h内逆转OGTAC-4对BRD4的修饰效果, 支持修饰的动态擦除, 适用于瞬时功能研究.
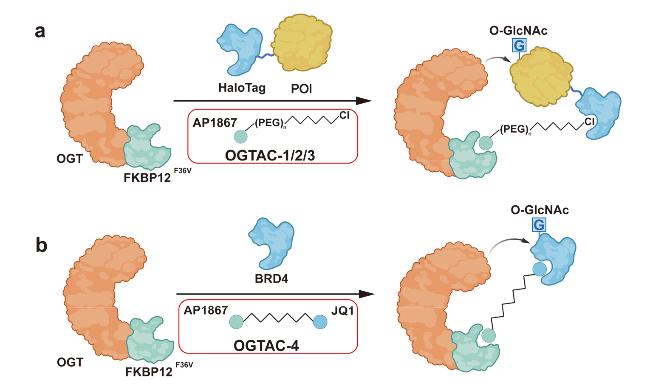
图9 靶向特定蛋白O-GlcNAc修饰的嵌合体分子. (a) FKBP12F36V融合表达的OGT通过OGTAC-1/2/3与融合表达HaloTag的靶蛋白结合, 催化靶蛋白发生O-GlcNAc修饰. (b) FKBP12F36V融合表达的OGT通过OGTAC-4与内源蛋白BRD4直接结合, 催化BRD4发生O-GlcNAc修饰Figure 9 Chimeric molecules for targeting O-GlcNAcylation to specific proteins. (a) OGT fused to FKBP12F36V is recruited to HaloTag-fused target proteins via OGTAC-1/2/3, resulting in O-GlcNAcylation of the target proteins. (b) OGT fused to FKBP12F36V directly binds the endogenous protein BRD4 through OGTAC-4, leading to O-GlcNAcylation of BRD4 |
最近, Xu等[47]又利用OGTACs技术探究了CK2α在Ser347位点的O-GlcNAc修饰对其稳定性、泛素化途径及下游信号通路的调控机制. 经过100 nmol/L的OGTAC-1处理8 h后, HTN-CK2α的O-GlcNAc修饰水平升高约2倍. 利用半乳糖基转移酶标记结合PEG化实验, 进一步证实Ser347修饰位点唯一性, 排除非特异性应激效应. 环己酰亚胺(CHX)追踪实验显示, OGTAC-1处理可加速HTN-CK2α降解, 该结果在HeLa和HCT116细胞中保持一致, 且蛋白酶体抑制剂MG132可挽救降解, 而溶酶体抑制剂氯喹则无效. 泛素化结果分析表明, OGTAC-1诱导的O-GlcNAc修饰使CK2α泛素化水平提升约1.5倍, 揭示O-GlcNAc修饰通过泛素-蛋白酶体系统(UPS)调控其稳定性. 机制上, CK2α被鉴定为E3泛素连接酶复合物CUL4A-DDB1-CRBN (CRL4CRBN)的底物. Co-IP实验证实CK2α与CRL4CRBN直接互作, 过表达复合物组分均可增强CK2α泛素化. OGTAC-1诱导的O-GlcNAc修饰进一步强化CK2α与CRBN的相互作用, 且该互作水平与O-GlcNAc修饰程度呈正相关, 表明O-GlcNAc修饰通过增强CRL4CRBN识别促进泛素化. 下游信号方面, O-GlcNAc修饰无法改变CK2α的激酶活性, 但是可以影响其底物识别的选择性, 重塑下游磷酸化网络. 经过OGTAC-1处理后, CK2α底物Akt在Ser129和Ser473位点的磷酸化水平显著提升, 以及PFKP在Ser386位点的磷酸化水平提升至约1.5倍, 该现象在CK2αS347A突变体中消失.
OGTACs为研究O-GlcNAc修饰在调控蛋白质功能中的作用提供了创新工具, 有助于揭示O-GlcNAc修饰驱动的全新调控机制. 在临床应用方面, OGTACs具有开发潜力, 可用于靶向与癌症及神经退行性疾病相关蛋白的异常O-GlcNAc修饰. 然而, OGTACs的应用目前仍依赖外源性系统, 需同时共表达带有HaloTag的目标蛋白与fOGT, 这在一定程度上限制了其对内源性蛋白的研究. OGTACs已在O-GlcNAc功能解析及精准治疗策略开发中展现出革命性潜力, 未来有望在基础生物学研究和转化医学实践中发挥重要作用.
2.6 基于双特异性RNA适配体的O-GlcNAc修饰调控
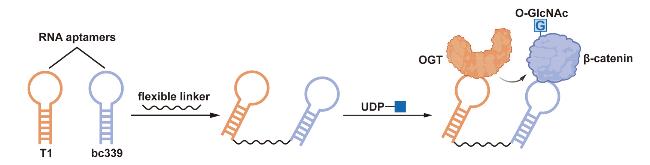
Zhu等[48]发表在《Cell》的研究中, 开发了一种基于双特异性RNA适配体(dual-specificity RNA aptamers, DS adapter)[49], 利用内源OGT和OGA调控特定蛋白O-GlcNAc 修饰水平的方法. 该研究通过指数富集系统进化技术(SELEX)[50], 筛选出靶向核质型OGT(ncOGT)的非抑制性适配体T1及靶向β-catenin的适配体bc339, 并将二者通过柔性连接子和折叠连接子相连, 构建了NL8F70、NL8F100和3JB8F+12, 在细胞中成功诱导OGT与β-catenin的空间位置接近, 从而特异性增强β-catenin的O-GlcNAc修饰, 且不影响全局O-GlcNAc水平或OGT/OGA表达(图10). 接下来Zhu等[48]又利用DS适配体探究了O-GlcNAc修饰如何调节β-catenin的功能. β-catenin作为转录因子却不具有DNA结合结构域, 其功能依赖于与其他蛋白质(包括EZH2、KAT2A和EP300)的相互作用[51-52]. 结果显示, 3JB8F+12通过增强β-catenin的O-GlcNAc修饰, 增强其与组蛋白甲基转移酶EZH2和乙酰转移酶KAT2A的相互作用, 抑制其与EP300/KAT5结合.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
然而, 若DS适配体持续结合在OGT及其底物蛋白上, 是否会干扰它们的正常功能是一个潜在问题. 因此, Zhu等通过整合核糖核酸开关元件设计了可诱导的DS适配体. 核糖核酸开关是位于mRNA上的非编码RNA元件, 能通过结合小分子配体改变自身构象, 进而调控基因表达[53]. 核糖开关可划分为三个功能模块, 负责配体结合的传感元件、执行下游效应的执行元件, 以及一个连接二者的传递序列[54]. 可诱导DS适配体LRS1F50C中整合了两个Mango核糖开关. 其中, 荧光Mango适配体作为传感元件, 其配体TO1-biotin是一种无毒、可透过细胞膜的小分子, 并能以低纳摩尔亲和力结合Mango[55]. T1和AP3适配体充当执行元件, TO1的加入可触发Mango折叠, 从而破坏T1和AP3的折叠, 使DS适配体功能“关闭”. 在细胞中进行验证, LRS1F50C使GFP-β-catenin的O-GlcNAc修饰水平提高了3倍, 且未影响内源β-catenin, 该效应随着TO1的加入被抑制. 此外, Zhu等[48]还采用Tet-On诱导表达系统来调控DS适配体的功能. 在该系统中, 适配体 3JB8F+12在U6WT启动子驱动下持续表达, atdT1则由融合了Tet 操作元件的U6TO启动子驱动. Doxycycline(Dox)可激活atdT1的表达, 从而干扰 3JB8F+12的折叠. 在细胞中, 该系统在无Dox条件下促进内源β-catenin的O-GlcNAc修饰, 而添加Dox 则终止该功能. 由于适配体在细胞内的半衰期较短, 可诱导表达系统能以高时间分辨率动态调控DS适配体的功能.
Zhu等[48]开发的DS aptamers提供了一种在不干扰内源OGT/OGA水平的前提下, 实现蛋白质O-GlcNAc修饰特异性调控的创新工具. 该策略通过灵活的连接策略、多样的调控机制(包括核糖开关和Tet-On系统)以及在细胞内的可逆控制能力, 不仅验证了β-catenin的O-GlcNAc修饰在调控其蛋白互作中的重要作用, 也为研究其他蛋白的O-GlcNAc修饰功能提供了通用化的技术平台. 该系统在未来可广泛应用于细胞信号通路解析、疾病机制研究及糖基化靶向治疗策略的开发.
2.7 位点特异性O-GlcNAc修饰蛋白的化学合成
随着高效肽段连接技术的发展, 研究人员现已经能够通过趋同的合成策略及高度化学选择性的连接反应, 将经过修饰的肽段精准拼接, 从而构建结构和功能上均接近天然状态的全长蛋白质.
早期研究主要集中在通过化学糖基化合成用于固相多肽合成(SPPS)的O-GlcNAc修饰氨基酸构建模块. 1995年, Meinjohanns等[56]报道了O-GlcNAc-Ser/Thr构建模块的合成方法. 该方法基于邻基参与策略, 利用Troc或Teoc保护的糖基供体与Fmoc-Ser/Thr活性酯(OPfp ester)在银催化条件下进行糖苷化反应, 成功获得具有β-构型的 O-GlcNAc修饰氨基酸单体, 可用于SPPS逐步拼接多肽链. 尽管该方法在当时具有重要开创意义, 但其合成路线较长, 涉及多步糖基保护、脱保护及对无水、无氧条件的严格要求, 操作门槛较高. 为解决传统方法的复杂性与普适性问题, De Leon等[57]于2018年提出了更加简便且高效的策略. 该方法利用商业化的β-Ac4GlcNAc与Fmoc保护的Ser、Thr或Cys在InBr3催化下直接发生糖苷化反应, 无需额外保护基调控, 也无需严格无水条件, 操作步骤大幅简化. 该反应同样依赖C2乙酰基的邻基参与机制, 从而实现高选择性地合成β-O-GlcNAc 及β-S-GlcNAc氨基酸构建模块. 最终产物可转化为OPfp酯, 兼容于常规SPPS条件, 可广泛用于糖肽及糖蛋白片段的构建.
虽然SPPS技术实现了高效构建携带O-/S-GlcNAc修饰的肽段, 但为了进一步整合这些带有位点特异性O-GlcNAc修饰的肽段, 研究者们发展了天然化学连接(native chemical ligation, NCL)技术. NCL是一种高化学选择性的肽段连接技术, 由Dawson及其同事[58]于1994年首次提出. 该技术在温和的水相条件(pH 7~7.5)下, 使肽段C端肽硫酯与N端半胱氨酸肽反应, 在两个片段之间生成天然肽键. 该反应首先生成硫酯键连接的中间体, 随后发生S→N酰基迁移, 最终生成以Cys为连接点的天然肽键产物. 然而, NCL的应用受到SPPS制备的肽段尺寸限制的制约[59]. 为此, 表达蛋白连接(expressed protein ligation, EPL)作为NCL的扩展方法应运而生[60]. 这种方法依赖于重组表达蛋白质片段, 这些片段要么带有C末端硫酯, 要么带有N末端半胱氨酸残基. 然后, 这些较大的蛋白质或蛋白质硫酯可以连接到通过SPPS合成的位点特异性O-GlcNAc修饰的肽段上. EPL技术允许在NCL中使用重组蛋白片段, 从而突破了多肽总长度的限制, 并扩展了可引入的修饰类型范围.
已有研究利用SPPS和EPL/NCL技术, 研究位点特异性O-GlcNAc修饰蛋白的功能. Shi等[61]通过合成肽微阵列技术和SPPS技术, 筛选并合成了ZO-3_357-371肽段, 用于研究O-GlcNAc糖基化修饰与磷酸化修饰之间的串扰调控. 结果表明Tyr364为ZO-3_357-371的磷酸化修饰位点, Ser369为O-GlcNAc修饰位点, 且Tyr364位点的磷酸化修饰可抑制附近Ser369位点的O-GlcNAc修饰, 而Ser369位点的O-GlcNAc修饰对在Tyr364处的磷酸化修饰没有显著影响, 表明Tyr磷酸化修饰可能对O-GlcNAc修饰的动态调节有显著贡献, 而O-GlcNAc修饰对磷酸化修饰信号传导的影响较小. 然而, 如果仅研究激酶和OGT的作用, 而不考虑由磷酸酶和OGA催化的逆向反应, 就无法全面理解这种串扰调控关系. Sharif等[62]的研究结果显示邻近位点的磷酸化修饰会影响OGA去除O-GlcNAc修饰, 但这种影响程度并不像其对OGT催化的O-GlcNAc修饰那样显著. 另一方面, 磷酸化和去磷酸化过程仅会受到邻近Ser上 O-GlcNAc修饰的轻微影响. Tarrant等[63]研究发现CK2催化亚基CK2α在Ser347位点存在O-GlcNAc修饰, 邻近细胞周期蛋白依赖性激酶磷酸化修饰位点Thr344. 随后通过EPL和SPPS技术进一步研究证明, Thr344位的磷酸化修饰通过与Pin1的相互作用提高了CK2α的细胞稳定性, 而Ser347的O-GlcNAc糖基化则与Thr344的磷酸化修饰拮抗, 并促进其通过蛋白酶体途径降解.
Schwagerus等[64]首次实现了携带位点特异性 O-GlcNAc修饰的tau蛋白半合成. 通过SPPS制备功能化肽段, 包括在Ser400位点含O-GlcNAc修饰的tau蛋白片段1和含光裂解生物素标签的tau蛋白片段2. 随后通过NCL将两个片段连接, 获得tau蛋白C端糖基化肽段. 经去硫反应在连接位点生成天然的丙氨酸后, N端半胱氨酸得以暴露, 该肽进一步通过EPL与其他片段结合, 最终生成全长tau蛋白, 并通过光可切除的生物素标签进行纯化. 该方法为研究O-GlcNAc修饰对tau蛋白的体外结构与功能调控提供了重要工具.
总体而言, SPPS与NCL/EPL等高效连接策略的结合, 不仅实现了位点特异性O-GlcNAc修饰氨基酸模块和修饰肽段的可控构建, 也为获得结构和功能均接近天然状态的全长修饰蛋白提供了可能. 这些方法学的进步为深入解析O-GlcNAc修饰在蛋白质折叠、稳定性、相互作用及疾病相关功能中的作用奠定了坚实基础, 并为未来系统性研究O-GlcNAc修饰蛋白的生物学机制开辟了新途径.
2.8 基于Tag‑and‑Modify策略的位点特异性O‑GlcNAc修饰
“标记与修饰”(Tag-and-Modify)方法通过将具有独特反应性的化学基团安装到蛋白质中(即“标记”), 然后对该基团进行选择性或特异性修饰[65]. 在天然残基中, Cys是最广泛使用的标记, 其具有高亲核性, 是亲电试剂选择性反应的经典靶点. 通过定点诱变可以轻松地将Cys安装在目标位点, 且其天然丰度相对较低, 通常能够制备单一Cys突变体. Chalker等[66]开发了一种将Cys转化为脱氢丙氨酸(Dha)的通用方法. 当Cys标记的蛋白经亲电性氮源O-间甲基苯磺酰羟胺(MSH)处理时, 会迅速发生消除反应生成Dha, 随后利用Dha作为反应性标签, 进一步获得多种翻译后修饰. N-乙酰氨基葡萄糖硫醇(GlcNAc-SH)带有一个巯基, 作为亲核试剂可以与Dha的双键发生Michael加成反应, 可在蛋白质侧链上引入GlcNAc, 形成O-GlcNAc修饰类似物S-GlcNAc修饰. 该策略已被用于研究O-GlcNAc修饰对组蛋白稳定性的调控[67]. 体外生产并纯化H2A重组的Thr101Cys突变体, 生成Cys101携带S-GlcNAc修饰的均一蛋白, 结果显示H2A-Thr101的S-GlcNAc修饰会破坏H2A/H2B二聚体的稳定性, 降低核小体整体稳定性, 从而促进染色质的开放. 同样的, 表达并纯化Ser112Cys的H2B, 并在Cys112位引入S-GlcNAc修饰, 结果表明H2B-Ser112的S-GlcNAc修饰并不影响核小体的组装, 但会改变其与蛋白质复合物的相互作用模式, 调控FACT染色质重塑复合物的结合.
然而, 该策略仍存在一定局限性. 首先, 反应产生的S‑GlcNAc修饰通过Cys的硫醚键与蛋白质侧链相连, 这并非天然的糖苷键, 因此只能作为天然O-GlcNAc修饰的模拟, 而其化学性质可能与天然修饰存在差异. 其次, 这类方法在形成Dha并进行亲核加成过程中, α-碳常发生外消旋化, 导致产物为不可分离的非对映异构体混合物, 这些异构体可能表现出不同的生物学特性, 从而对实验结果的解释带来一定的不确定性.
3 位点特异性O-GlcNAc修饰在疾病中的作用
3.1 神经退行性疾病
3.1.1 阿尔茨海默症
O-GlcNAc修饰与磷酸化修饰在tau蛋白上呈现动态拮抗关系. Liu等[73]证明, 在使用OGA抑制剂PUGNAc处理过表达tau蛋白的PC-12细胞时, tau蛋白上的O-GlcNAc修饰水平增加, 而其磷酸化修饰水平则以位点特异性的方式下降. Yuzwa等[74]开发的强效OGA抑制剂Thiamet-G能够使O-GlcNAc修饰水平升高, 并在PC-12细胞以及体内(大鼠大脑皮层和海马体)中阻断tau蛋白在相关的病理位点(T231、S396和S422)处的磷酸化. 在低葡萄糖条件下, 可观察到tau蛋白上特定位点O-GlcNAc修饰水平降低而磷酸化修饰水平增加, 使用PUGNAc处理挽救O-GlcNAc修饰水平, 能够抑制tau蛋白的过度磷酸化. Liu等[72]又通过向大鼠脑内注射DON(一种GFAT抑制剂)抑制HBP途径, 降低大鼠大脑中O-GlcNAc修饰水平, 同时tau蛋白在某些位点的磷酸化增加. Liu等[72]通过在HEK293细胞中敲低OGT进一步证明, O-GlcNAc修饰水平的降低与tau蛋白磷酸化水平的升高密切相关, 尤其是在T205和T212这两个磷酸化位点.
综上所述, O-GlcNAc修饰通过拮抗tau磷酸化、抑制聚集、激活自噬及调控APP代谢等多重机制参与AD病理进程. 尽管其治疗潜力显著, 但需深入解析不同生理背景下O-GlcNAc的动态平衡及其与其他信号通路的交互作用, 以开发精准且安全的AD干预策略.
3.1.2 帕金森症
帕金森症(Parkinson’s disease, PD)是第二常见的神经退行性疾病, 影响患者的认知、运动功能、睡眠等多个方面的功能[80]. PD的病理特征与α-突触核蛋白的聚集密切相关, 研究表明, α-突触核蛋白存在O-GlcNAc糖修饰[81-82]. α-突触核蛋白的中央非淀粉样β成分(NAC)区域对于α-突触核蛋白的聚集至关重要, 且多项研究证实该区域的T72、S87等位点存在O-GlcNAc修饰[83]. Marotta等[84]利用EPL技术制备了T72位点特异性O‑GlcNAc修饰的α‑突触核蛋白. 结果显示, 与野生型α‑突触核蛋白相比, O‑GlcNAc修饰的α‑突触核蛋白未形成任何二级结构和纤维状聚集体. O‑GlcNAc修饰阻断了α‑突触核蛋白的寡聚化, 且不影响其与膜结合形成α-螺旋的正常功能. Lewis等[85]的研究则发现, S87位点的O-GlcNAc修饰虽能减缓聚集动力学, 但其效果弱于T72位点O-GlcNAc修饰或S87E磷酸模拟突变, 磷酸化会破坏蛋白的膜结合功能, 而O-GlcNAc修饰则保留了这一内源活性, 凸显其病理调控中的独特优势.
机制上, O-GlcNAc修饰通过多重途径抑制α-突触核蛋白聚集. 分子动力学模拟显示, T81单位点修饰或T72/T75/T81三联位点修饰通过糖基的空间位阻效应干扰单体间氢键网络的形成, 削弱α‑突触核蛋白寡聚 化[86]. 构象分析表明, T72位点O-GlcNAc修饰使蛋白结构更松散, 而S87位的O-GlcNAc修饰则诱导紧凑构象, 两者均延缓α‑突触核蛋白纤维形成[14]. 此外, O-GlcNAc修饰可抑制钙蛋白酶对α-突触核蛋白的分解, 减少促聚集性片段的产生[87]. Zhang等[88]的研究进一步揭示, O-GlcNAc修饰促进蛋白形成SDS抗性寡聚体, 可能通过稳定非纤维化构象抑制病理聚集. 研究还发现, O-GlcNAc修饰对α-突触核蛋白的调控具有位点依赖性. T75单位点修饰或T72/T75/T81三联位点修饰可阻断体外纤维延伸及神经元内聚集体的毒性, 而T72单位点修饰在体外并未阻止聚集体的延伸[81]. 此外, O-GlcNAc对其他分子伴侣的修饰也间接影响α-突触核蛋白稳态. HSP27、α-晶状体蛋白等小热休克蛋白(sHSPs)的O-GlcNAc修饰可增强其抗淀粉样活性, 通过阻断自身结构域内相互作用, 促进与错误折叠蛋白的结合, 从而抑制聚集. HSP27的O-GlcNAc修饰可增强其与BAG3/HSP70复合物的协同作用, 提升对模型底物的再折叠能力[89]. 在治疗策略探索中, OGT过表达或OGA抑制剂(如Thiamet-G、ASN90)处理可升高O-GlcNAc修饰水平, 减少α-突触核蛋白磷酸化及病理聚集, 改善PD模型小鼠的运动功能[90-91]. 但需注意, O-GlcNAc过度升高可能抑制自噬, 导致蛋白清除障碍, 提示需精准调控其动态平衡. 最新研究还发现, O-GlcNAc修饰的α-突触核蛋白纤维(PFFs)结构不同于病理性纤维, 其种子效应较弱且毒性降低, 或为干预病理扩散提供新思 路[92].
综上, O-GlcNAc修饰通过直接抑制α-突触核蛋白聚集、调控分子伴侣功能及细胞应激通路, 形成多维度抗PD机制. 尽管其双重效应需进一步平衡, 但针对O-GlcNAc的动态调节仍是极具前景的治疗方向, 为神经退行性疾病的干预开辟了新路径.
3.1.3 夏科-马里-图斯病
Moon等[95]基于先前O-GlcNAc修饰与HSP27活性之间的研究, 在CMT2型(CMT2)疾病背景下进一步探究二者关联. CMT2患者可能携带突变的HSP27, 导致分子伴侣活性受损, 进而引发疾病. Moon等通过半合成策略构建了在T184糖基化位点突变的HSP27蛋白, 并测试其对抗β淀粉样蛋白聚集的分子伴侣活性. 研究发现, O-GlcNAc修饰能够“挽救”可形成寡聚体的HSP27突变体, 而对其他突变体, O-GlcNAc修饰反而导致蛋白不稳定. 尽管造成这些差异的机制尚不清楚, 但该研究表明O-GlcNAc修饰对HSP27稳定性的影响依赖于其氨基酸序列, 特定位点突变可能通过改变修饰位点的微环境引发功能代偿或紊乱.
另一方面, 针对NF-L组装调控的最新研究进一步揭示了O-GlcNAc修饰的位点特异性效应. 通过OGT过表达及OGA/OGT抑制剂干预, 研究者在人NF-L头部结构域中鉴定了T21、S27、S34和S48四个O-GlcNAc修饰位点. 提高O-GlcNAc修饰水平可显著增加NF-L的可溶性, 而单个Ser/Thr残基突变为Ala(如T21A)的突变体也表现出类似效应. 值得注意的是, 当四个修饰位点同时突变(NF-L4A)时, 则蛋白形成为类似野生型的不可溶性丝状结构, 提示多位点O-GlcNAc修饰通过协同作用动态平衡NF-L的溶解-组装状态. 随后通过光亲和交联实验, 研究者进一步证实O-GlcNAc修饰直接介导NF组装过程中与其他蛋白的相互作用, 而CMT相关NF-L突变体中异常的O-GlcNAc修饰模式可能通过干扰此类互作导致病理表型[96].
综合上述研究, O-GlcNAc修饰通过位点特异性和序列依赖性机制, 在CMT疾病中形成复杂的调控网络, 一方面动态调节HSP27的分子伴侣活性与稳定性, 另一方面精准调控NF-L的组装状态及相互作用. 这些发现不仅深化了对CMT病理机制的理解, 也为开发基于蛋白质翻译后修饰调控的新型疗法奠定了理论基础.
3.2 肿瘤
在肿瘤微环境中, 高葡萄糖通量提升全局O-GlcNAc修饰水平, 重塑肿瘤细胞能量供应格局. 磷酸果糖激酶-1(PFK1)的S529位O-GlcNAc修饰通过空间位阻抑制其与果糖-2,6-二磷酸(F-2,6-BP)结合, 破坏PFK1四聚体稳定性并降低酶活性, 促使糖酵解通量转向磷酸戊糖途径(PPP), 增加NADPH和谷胱甘肽(GSH)合成以增强抗氧化能力[107]. 丙酮酸激酶M2型(PKM2)的多位点 O-GlcNAc修饰(T405/S406、S362/T365)破坏其四聚体组装, 抑制其酶活性并促进核转位, 并可以通过上调GLUT1和LDHA表达来强化Warburg效应[108]. 转移性结直肠癌细胞(SW620)中PKM2 的O-GlcNAc修饰水平高于非转移性细胞(SW480)[109]. PKM2的S333A突变小鼠模型显示加速肿瘤生长[110]. 类似地, 磷酸甘油酸激酶1(PGK1)T255位O-GlcNAc修饰增强其底物结合能力, 促进糖酵解和丝氨酸合成, 同时抑制TCA循 环[111].
肿瘤细胞基因组不稳定, 这使肿瘤细胞对DNA损伤更加敏感, 更依赖DNA损伤修复通路. 已有研究发现, O-GlcNAc修饰参与调控DNA修复酶的功能, 在DNA损伤修复中发挥重要作用. DNA聚合酶η(Polη)T457位的O-GlcNAc修饰促进其泛素化降解, 影响其跨损伤合成能力[112]. 酸性核质DNA结合蛋白1(AND-1)在S575和S893位点存在O-GlcNAc修饰, 在结直肠癌细胞中, 这些修饰被证实可以使细胞对电离辐射具有抵抗力[113]. Flap核酸酶1(FEN1)在暴露于基因毒性试剂时, 在S352位点发生动态O-GlcNAc修饰, 破坏该修饰的动态平衡会干扰DNA复制与DNA损伤响应, 使细胞对应激试剂更加敏感[114]. 拓扑异构酶IIα(TOP2A)S1469位的O-GlcNAc修饰增强其染色质结合及DNA双链断裂修复效率[115]. 聚(ADP-核糖)糖苷酶S26位的O-GlcNAc修饰促进PARG向染色质招募, 增强其在DNA损伤修复中的功能[116]. 转录因子NRF1(T342/T500位)的O-GlcNAc修饰可以稳定该蛋白, 并促进激活基因毒性应激反应基因的转录[117], MTA1(S237/S241/S246位)的O-GlcNAc修饰促进应急适应基因的转录, 帮助肿瘤细胞适应阿霉素造成的DNA损伤应激[118].
肿瘤细胞化疗耐药性的产生也与O-GlcNAc修饰密切相关. MITF的S49位O-GlcNAc修饰抑制其胞质滞留, 增强核转位并介导乳腺癌细胞对CDK4/6抑制剂Palbociclib的耐药[119]. 纤维蛋白(FBL)S142位的O-GlcNAc修饰能够促进肿瘤细胞的增殖, 以及小鼠模型中肿瘤的形成, 过表达野生型FBL可降低MCF-7细胞对阿霉素的敏感性[120]. SNARE蛋白降低的O-GlcNAc修饰水平通过促进外泌体分泌和细胞自噬这两种方式, 参与卵巢癌中顺铂耐药性的产生[121]. p53和c-MYC的O-GlcNAc修饰水平升高与肺癌顺铂耐药有关[122]. 最近的一项糖蛋白组学研究发现, 钙结合蛋白calreticulin(CRT)在T346位点的O-GlcNAc修饰能降低顺铂诱导的内质网应激和细胞死亡[123]. 因此, 靶向O-GlcNAc修饰在化疗耐药中的调控机制, 有望成为将O-GlcNAc修饰调控剂与传统化疗药物联合使用的治疗策略基础.
肿瘤细胞的转移与侵袭过程O-GlcNAc修饰也参与调控. Cofilin的S108位O-GlcNAc修饰促进侵袭伪足形成, 增强肿瘤细胞迁移[124]. 控制EMT相关基因表达的蛋白也受O-GlcNAc修饰调控, HDAC1在Thr114和Ser263位的O-GlcNAc修饰与促进肿瘤细胞增殖及降低上皮标志物E-cadherin表达相关, 转化生长因子β(TGF-β)信号促进MCF-7细胞中染色质重塑因子MORC2的Thr556位点O-GlcNAc修饰, 这一修饰与细胞增殖和EMT相关基因SNAIL与CTGF的转录上调相关[125-126]. 在乳腺癌患者样本中, 较低的OGT和MORC2水平与更好的预后相关[125]. STAT3的T717位O-GlcNAc修饰在缺氧条件下稳定蛋白并激活EMT相关基因[127]. 深入研究 O-GlcNAc修饰如何调控肿瘤细胞恶性表型, 将有助于更好地理解EMT的生物学机制, 并为这些高度侵袭性的肿瘤寻找潜在的治疗靶点.
3.3 糖尿病
糖尿病以持续性高血糖和能量代谢紊乱为核心特征[128-129]. 细胞内长期除处于高糖状态增强了HBP途径通量, 并最终影响O-GlcNAc修饰水平. 大量研究表明, O-GlcNAc修饰的失调与2型糖尿病密切相关[130]. 胰岛素抵抗是2型糖尿病的重要发病机制之一[131]. O-GlcNAc修饰水平或O-GlcNAc相关通路的异常均会导致胰岛素抵抗表型出现[132-133]. 在多种细胞和啮齿类动物模型中, 增加氨基葡萄糖[134]、过表达HBP途径限速酶GFAT[135]、提高OGT表达水平[136], 或降低OGA表达水平[137], 均与胰岛素抵抗的发生直接相关. 然而也有研究发现, 抑制HBP途径或敲低OGT同样会引起胰岛素分泌缺陷[138-139]. 这提示无论是升高还是降低O-GlcNAc修饰水平, 均与糖尿病表型的形成相关. 因此, O-GlcNAc修饰的精细调控是维持正常细胞稳态的重要因素, 其失衡与糖尿病的发生发展密切相关.
越来越多证据表明, 糖尿病相关的分子功能异常并非源于全局O-GlcNAc修饰, 而是由特定位点修饰模式的选择性重塑所驱动. 胰岛素信号通路包含多种核心蛋白, 如蛋白激酶B(AKT)、转录因子FOXO1、蛋白酪氨酸磷酸酶(PTP1B)、胰岛素受体底物(IRS)等, 这些蛋白均被报道存在O-GlcNAc修饰, 这提示O-GlcNAc修饰可能在糖尿病状态下介导胰岛素信号的失调. Wu等[140]的研究发现, 在高葡萄糖环境下, 肝细胞中OGT表达上调, 进而催化蛋白激酶B1(AKT1)在Thr423和Ser466位点发生O-GlcNAc修饰. 该修饰显著提高了AKT1的稳定性, 并增强了其与FOXO1的相互作用. O-GlcNAc修饰的AKT1进一步催化FOXO1的Ser238位点发生磷酸化修饰, 从而阻止FOXO1核易位, 使其滞留于细胞质中. 这使FOXO1无法结合并激活糖噬关键基因GABARAPL1的启动子, 导致GABARAPL1及其介导的糖噬过程被显著抑制. 与此同时糖原合成相关基因表达上调, 最终导致肝细胞中糖原的异常积累. 该通路完整揭示了高葡萄糖通过OGT-AKT1-FOXO1通路调控糖原代谢的新机制, 为理解2型糖尿病患者肝脏糖代谢异常及胰岛素抵抗提供了新思路. Housley等[141]发现在肝脏细胞中FOXO1存在O-GlcNAc修饰, 其修饰水平在链脲佐菌素诱导的糖尿病大鼠模型中显著增强. 随后该研究通过ETD MS/MS技术精确鉴定了FOXO1的四个O-GlcNAc修饰位点: Thr317、Ser550、Thr648和Ser654. 其中Thr317位点的功能尤为重要, 该位点的突变显著削弱了葡萄糖诱导的FOXO1转录激活能力. 在功能层面, O-GlcNAc修饰的FOXO1可上调糖异生基因(Pepck、G6pc)和氧化应激响应基因(catalase、MnSOD)的表达, 提示O-GlcNAc修饰通过调控FOXO1的转录活性, 将高糖信号与糖异生及氧化应激应答耦联, 为糖尿病中代谢失衡的发生提供了明确的分子机制解释. 另外, FOXO1在T314、S547、S645和S651位点的O-GlcNAc修饰与神经生成素3(Ngn3)基因转录抑制增强相关, 从而阻断肠内分泌祖细胞分化为产生胰高血糖素样肽-1 (GLP-1)的L细胞[142]. 该研究提示, 可通过干预调控O-GlcNAc修饰水平为2型糖尿病治疗及GLP-1分泌调节提供新的思路. PTP1B作为一种胰岛素信号负调控因子, 在胰岛素抵抗中发挥关键作用. 在胰岛素抵抗模型中, PTP1B的O-GlcNAc修饰水平显著升高. 研究发现, PTP1B存在三个O-GlcNAc修饰位点(Ser104、Ser201和Ser386), 其中Ser104和Ser201位于其磷酸酶催化结构域内部. 通过定向位点突变(S104A或S201A)降低PTP1B的O-GlcNAc修饰, 削弱其磷酸酶活性, 从而减轻其对胰岛素信号的抑制作用, 恢复胰岛素刺激下的Akt和GSK3β磷酸化水平[143]. 提示PTP1B特定位点的O-GlcNAc修饰在胰岛素抵抗的发生中具有关键调控作用. 此外, Kaleem等[144]的研究表明, 当UDP-GlcNAc水平增加时, IRS-1的Ser1101位点和IRS-2的Ser1149位点发生O-GlcNAc修饰, 这可能促进胰岛素抵抗表型的出现. 而当IRS-1的Ser1101位点和IRS-2的Ser1149位点发生磷酸化时, 胰岛素信号传导则会受到抑制.
综上所述, O-GlcNAc修饰在糖尿病中的作用远不仅仅是全局性的. 越来越多的证据表明, 特定位点的O-GlcNAc修饰在糖尿病的代谢失衡和胰岛素抵抗的发生中起着关键作用. 特别是在肝脏和肠道内分泌细胞的功能调控中, O-GlcNAc修饰的位点特异性变化可能是糖尿病发生的关键驱动因素. 因此, 精细调控这些位点特异性的O-GlcNAc修饰, 不仅为糖尿病的机制研究提供了新的视角, 也可能为其治疗策略的创新提供宝贵的线索和靶点.
4 总结与展望
O-GlcNAc修饰作为一种十分重要的动态可逆的蛋白质翻译后修饰, 能够快速响应营养、能量状态和环境刺激, 从而精细调控蛋白质的折叠、稳定性、亚细胞定位、相互作用以及活性. 在信号转导、转录调控、染色质重塑、细胞周期和应激应答等关键生命过程中, 都扮演十分重要的角色. O-GlcNAc修饰水平的异常改变与多种疾病密切相关, 如在神经退行性疾病中影响蛋白聚集, 在糖尿病中参与胰岛素信号通路失衡, 在癌症中调节代谢重编程和细胞增殖.
之前的研究多集中于细胞或组织的整体 O-GlcNAc修饰水平, 探究其全局分布和总体变化趋势对关键生命过程的影响. 近年来研究者开始更加关注单个特定蛋白的O-GlcNAc修饰水平. 聚焦于对特定蛋白特定位点的精确调控不仅有助于揭示O-GlcNAc修饰在信号传导、基因表达调控及疾病发生发展中的具体作用, 也为我们理解其在细胞功能中的分子机制提供了更深入的视角. 由此, O-GlcNAc 研究从全局性观察逐步迈向靶向性解析. 近年来, 多种研究工具和技术的不断涌现, 从基因编辑、纳米抗体、RNA适配体, 到固相多肽合成、翻译后突变等化学方法, 研究者能够更精确地解析O-GlcNAc修饰的功能和意义. 这些技术的应用不仅揭示了位点特异性O-GlcNAc修饰在神经退行性疾病、肿瘤等重大疾病中的潜在作用, 也为发展靶向性更强的新型治疗策略奠定了基础.
然而, 现阶段的技术方法仍存在明显局限. 多数策略目前只能在体外体系中实施, 在细胞或动物体内仍无法直接应用. 其次, 即便有少数技术方法实现了内源环境下的尝试, 也往往需要借助标签融合蛋白或人工修饰的辅助系统, 这与细胞内生理环境存在差异, 不能完全代表O-GlcNAc修饰的真实调控状态. 这些局限性阻碍了我们对O-GlcNAc修饰动态调控作用的全面理解, 也在一定程度上影响了潜在治疗策略的转化应用.
未来随着多种技术不断发展, O-GlcNAc修饰研究有望克服现阶段体外体系依赖、体内应用受限等局限, 实现对内源性蛋白的精准调控. 将进一步延伸从全局水平到单蛋白、甚至位点特异性解析的研究视角, 使我们能够更加深入地揭示O-GlcNAc修饰的重要功能. 未来的研究不仅有望阐明O-GlcNAc修饰在疾病发生发展中的具体分子机制, 还可能将其作为疾病生物标志物或精准干预靶点, 为相关疾病的诊断与治疗开辟新的途径.
(Cheng, B.)