


1 Introduction
Alkyl carboxylic acids are among the most abundant and versatile organic compounds in nature. They are cheap, highly stable, non-toxic, and widely available for use in organic synthesis.[1] In recent years, significant progress has been made in utilizing carboxylic acids or their redox-active esters (RAEs) as radical precursors for decarboxylative cross-coupling reactions in the presence of transition-metal catalysts or photocatalysts,[2] such as decarboxylative halogenation,[3] decarboxylative C—C cross-coupling reactions,[4] decarboxylative borylation,[5] and others.[6] Among these, decarboxylative amination[7] has garnered increasing attention for the synthesis of alkyl amine, which is important structural motif in pharmaceuticals, agrochemicals, and organic materials. For example, in 2017, Fu, Peters, and co-workers[8] disclosed the first example of a photoinduced copper-catalyzed intramolecular decarboxylative C—N coupling of alkyl RAEs to generate protected amines. This reaction represents an alternative to the Curtius rearrangement but is limited to primary and secondary alkyl RAEs. Subsequently, Hu and co-workers[9] developed an elegant intermolecular coupling of alkyl RAEs with benzophenone imines by merging Ir-based photoredox with copper catalysis (Scheme 1a). This method successfully converted primary, secondary, and tertiary alkyl carboxylic acids, providing an effective protocol for the synthesis of structurally diverse primary amines. Additionally, the Lopchuk[10] and Shang[11] groups also reported different decarboxylative amination methods using alkyl RAEs as radical precursors to construct protected amines through thermochemical (Scheme 1b) and photochemical (Scheme 1c) pathways, respectively. Despite these achievements, most of these methods depend on photoredox or transition metal catalysts. Therefore, the development of a mild and straightforward protocol for decarboxylative amination remains highly desirable, especially those enabled by metal- free catalysis.
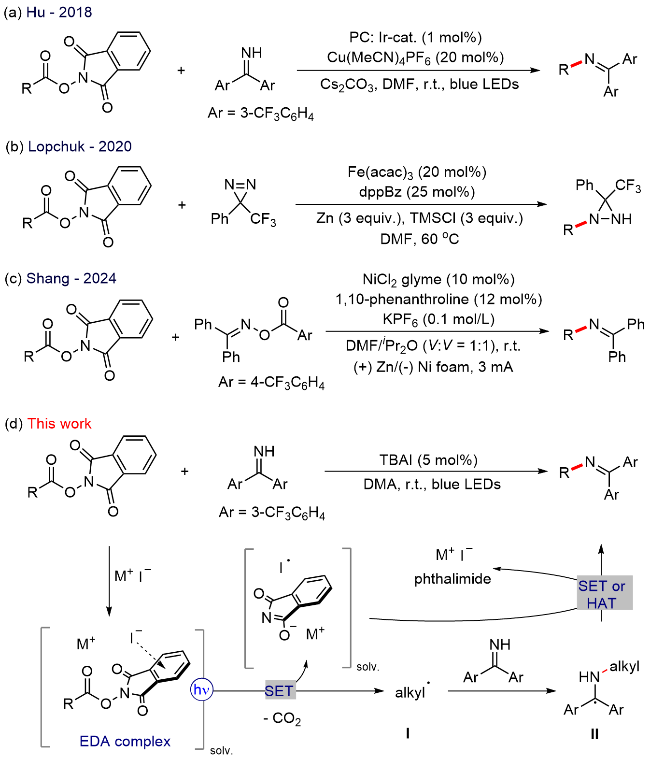
Recently, electron donor-acceptor (EDA) complex chemistry has garnered extensive attention in the field of synthetic photochemistry.[12] It offers a valuable alternative for generating radical species under mild conditions without the need for transition-metal or organic dye-based photoredox catalysts. Our recent works revealed that iodide anions and triphenylphosphine, in combination with alkyl N-hydroxyphthalimide (NHPI) esters, can form an EDA complex that absorbs visible light to generate alkyl radicals.[13] Since then, this strategy has been successfully applied to construct a variety of C—C bonds by us[14a,14b] and other groups.[15]
Building on our ongoing research in EDA complex chemistry, we envisioned that such strategy could be extended to decarboxylative amination, thereby avoiding the use of transition metal catalysts in the construction of C—N bonds. The mechanistic details of our proposed decarboxylative amination are outlined in Scheme 1d. A combination of alkyl NHPI esters and iodide salts may effectively undergo an intermolecular single electron transfer (SET) through an anion-π interaction within a solvent cage under visible light irradiation, generating alkyl radicals (I).[14c] These alkyl radicals then undergo radical addition with benzophenone imines to smoothly form benzyl radical intermediates (II).[16] Subsequently, these benzyl radical intermediates can be oxidized by iodide radicals to produce benzyl cations,[13] which are then deprotonated by phtha- limide anions to yield the desired product. Additionally, another possible reaction pathway involves hydrogen atom transfer (HAT) between intermediate II and iodine radicals to facilitate product formation. This process is accompanied by the regeneration of iodide ions, completing the redox cycle.
2 Results and discussion
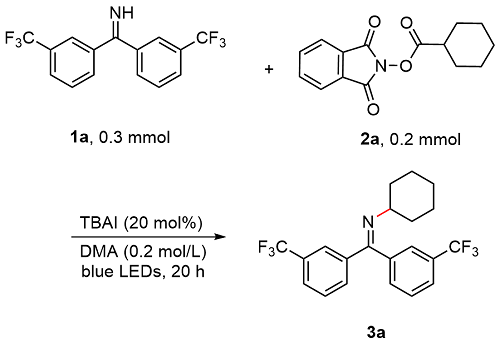
After carefully examining the feasibility of the proposed decarboxylative amination enabled by EDA complex with TBAI (tetrabutylammonium iodide) as an electron donor, a comprehensive investigation of all reaction parameters was conducted. We were delighted to find that when a solution of benzophenone imine 1a (1.5 equiv.) and cyclohexyl NHPI ester 2a (1.0 equiv.) in N,N-dimethylacetamide (DMA, 0.2 mol/L) was irradiated with blue light-emitting diodes (LEDs), 70% yield of the protected alkyl amine product 3a was obtained using just catalytic amounts of TBAI (Table 1, Entry 1). It was observed that reducing the amount of TBAI to 5 mol% still allowed for the formation of 73% 3a (Entry 2), but a lower yield was obtained upon increasing the amount of TBAI to 30 mol% (Entry 3). Solvation heavily affects the reaction outcome by influencing formation of a transiently assembled EDA encounter complex in the solvent cage. The yield of 3a was significantly lower when using N,N-dimethylformamide (DMF), N-methyl pyrrolidone (NMP), or acetone as the solvent (Entries 4~6). Furthermore, other iodide salts, including lithium iodide, sodium iodide, and potassium iodide, were found to be effective catalysts for providing 3a, albeit in lower yields compared to TBAI (Entry 7). Exploring different substituted benzophenone imines as the imine partners (Entries 8~10) revealed that the coupling of the electron-rich imines 1b~1d resulted in lower yields (30%, 28%, and 37%, respectively) than that of the electron-deficient imine 1a. This observation underscores the favorable nature of electron-deficient imines in the radical addition step. Finally, control experiments demonstrated that both light and iodide salts were essential for the success of the decarboxylative amination (Entry 11).
Table 1 Optimization of the reaction conditionsa |
| Entry | Variations from the above conditions | Yielda/% |
|---|---|---|
| 1 | None | 70 |
| 2 | 5 mol% TBAI | 73 |
| 3 | 30 mol% TBAI | 61 |
| 4 | DMF instead of DMA | 63 |
| 5 | NMP instead of DMA | 50 |
| 6 | Acetone instead of DMA | 14 |
| 7 | LiI, NaI, or KI instead of TBAI | 33~61 |
| 8 | 1b instead of 1a | 30 |
| 9 | 1c instead of 1a | 28 |
| 10 | 1d instead of 1a | 37 |
| 11 | No light (50 ℃) or TBAI | 0 |
|
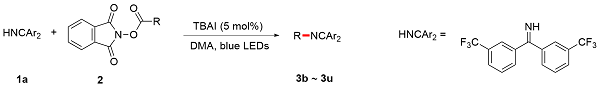
With the optimized conditions in hand, we then focused on exploring the scope of this EDA complex-driven decarboxylative C(sp3)—N bond-forming reaction. As depicted in Table 2, a diverse range of alkyl NHPI esters derived from primary, secondary, and tertiary carboxylic acids proved to be amenable substrates. Both acyclic (3b, 3c, 3l~3p) and cyclic (3d~3k) protected amines were obtained in moderate to good yields. Furthermore, the intra- and terminal olefin motifs (3h, 3i, 3p, 3t), which were highly susceptible to free radicals, were well tolerated, thus demonstrating the excellent chemical selectivity of this method. The NHPI ester derived from biomass-based tetrahydrofuroic acid was also a suitable substrate in this protocol (3j). Interestingly, for primary alkyl NHPI esters, 53% yield of the intramolecular decarboxylative C—N coupling product (3q) was successfully obtained in the absence of benzophenone imine.[8] Unfortunately, NHPI esters derived from amino acids were inactive in this protocol. Notably, the late-stage functionalization of derivatives from natural products and drug molecules, including gemfibrozil (3r), abietic acid (3s), and clofibrate (3t), proceeded smoothly to form the protected amine derivatives, showcasing the potential applications of this metal-free method in drug discovery endeavors.
Table 2 Scope of alkyl NHPI esters |
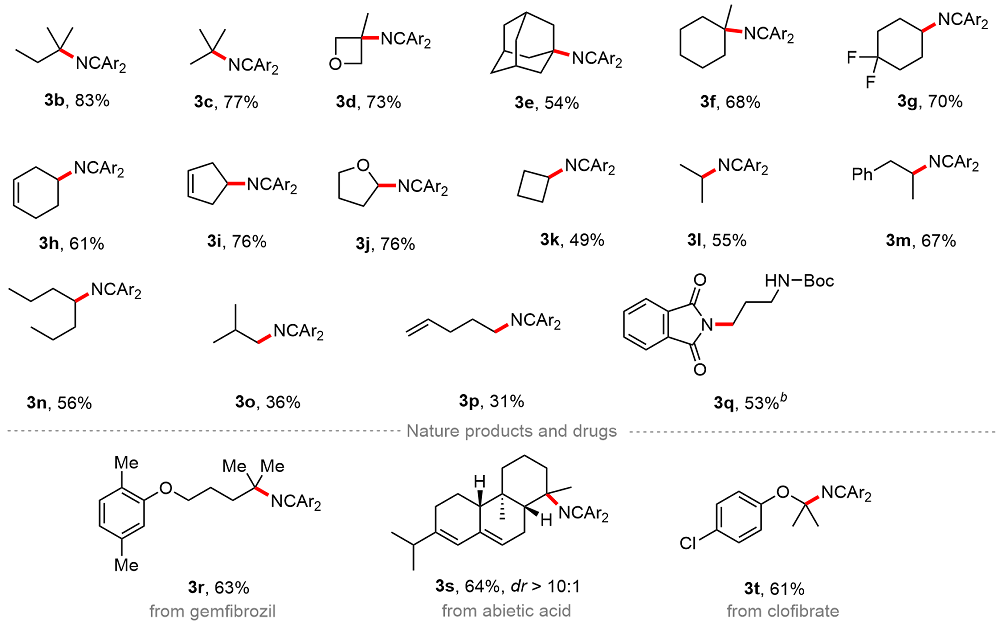 |
a Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), NHPI esters 2 (0.2 mmol, 1.0 equiv.), TBAI (5 mol%), DMA (0.2 mol/L), stirred at room temperature for 20 h under blue LEDs (450 nm, 18 W) irradiation. Isolated yields. b In the absence of 1. |
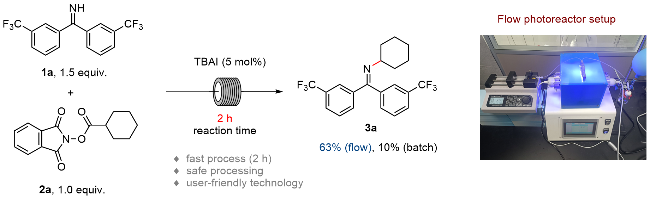
To further demonstrate the synthetic utility of this method, this decarboxylative amination in a continuous- flow process was conducted (Scheme 2). Gratifyingly, the efficient of this protocol was significantly improved in continuous-flow reactors compared to the batch reaction process. The simplicity and low cost of this catalytic system make it appealing for large-scale syntheses.[17]
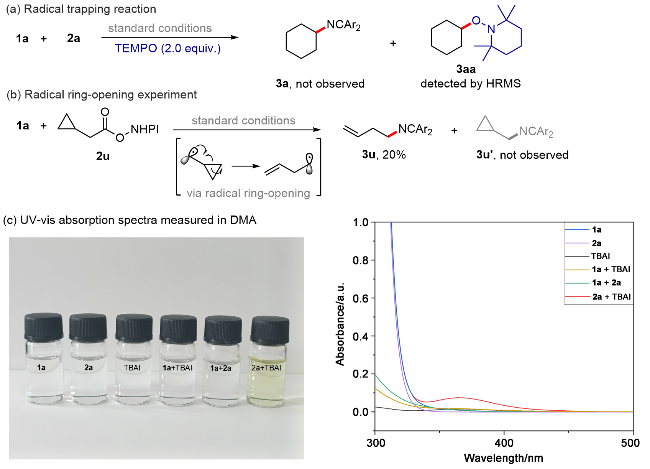
A series of mechanistic experiments were conducted to better elucidate the reaction mechanism. Initially, the radical trapping reaction showed that the formation of 3a was completely inhibited in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical scavenger. The TEMPO-adduct 3aa was successfully detected in the reaction mixture (Scheme 3a). Additionally, coupling of the NHPI esters derived from cyclopropylacetic acid 2u, which served as radical-clock probe, resulted in the ring-opening product 3u (Scheme 3b). These results strongly confirmed the intermediacy of a free alkyl radical in this transformation.[14b] Finally, UV-vis absorption spectra of the individual reaction components, benzophenone imine 1a, cyclohexyl NHPI ester 2a, TBAI, and their mixed solution, were measured in DMA (Scheme 3c). As expected, TBAI alone showed no absorption, while benzophenone imine and cyclohexyl NHPI ester exhibited absorption bands in the near-ultraviolet range. The combination of cyclohexyl NHPI ester with TBAI in DMA displayed a noticeable red shift, indicative of the formation of an intermediary EDA complex between cyclohexyl NHPI ester and TBAI. These mechanistic experiment results support, to some extent, the mechanism we proposed in Scheme 1d.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In summary, we herein report a photoinduced decarboxylative amination of alkyl NHPI esters with benzophenone imines without the need for transition-metal catalysts or photocatalysts. Derivatives from primary, secondary, and tertiary carboxylic acids proceed smoothly to afford a diverse array of acyclic and cyclic protected amines. Preliminary mechanistic studies indicate that this process is driven by the photoactivity of EDA complexes assembled from iodide salts with alkyl NHPI esters. The mild transition- metal-free reaction conditions, the capability for late-stage functionalization of complex molecules, and the successful implementation in continuous flow process are attractive features that underscore the synthetic potential and value of our methodology.
4 Experimental section
4.1 General method
The NMR spectra were recorded on a Bruker 600 MHz spectrometer and an Agilent 600 MHz spectrometer. The chemical shifts (δ) in 1H NMR were reported relative to tetramethylsilane (Me4Si) as internal standard (δ 0.0) or proton resonance resulting from incomplete deuteration of NMR solvent: CDCl3 (δ 7.26). 13C NMR spectra were recorded at 151 MHz, and the chemical shifts (δ) were reported relative to CDCl3 (δ 77.10). 19F NMR spectra were recorded at 564 MHz. The absorption spectra in solution were recorded on a UNIC 4802 UV/VIS double beam spectrophotometer in a 1.0 cm length quartz cell, all compounds were dissolved in DMA and measured at 25 ℃. HRMS analysis was performed on a Finnigan LCQ advantage Max Series MS System. ESI-mass data was acquired using a Thermo LTQ Orbitrap XL Instrument equipped with an ESI source and controlled by Xcalibur software.
4.2 General procedure for decarboxylative amination
Redox active esters (0.2 mmol, 1.0 equiv.) (if solid) and TBAI (5 mol%) were added in 10 mL Schlenk tube equipped with a stirring bar. The tube was evacuated and filled with argon (repeated for three times). To this solid, bis(3-(trifluoromethyl)phenyl)methanimine (0.3 mmol, 1.5 equiv.) and anhydrous DMA (1 mL) were added using a gastight syringe under argon atmosphere. The reaction mixture was stirred under irradiation with 450 nm blue LEDs (18 W), maintained at approximately room temperature in the air-conditioned room of 25 ℃. After 20 h, the resulting reaction mixture was directly purified by column chromatography (notably, the silica gel used for the purification was pre-neutralized with 5% triethylamine in hexanes solution prior to the usage, to minimize product loss).
4.3 Procedure of continuous-flow reactions
A 100 mL round bottom flask equipped with a magnetic stir bar was charged with 1,3-dioxoisoindolin-2-yl cyclohexanecarboxylate (5.0 mmol, 1.0 equiv.), bis(3-(trifluoro-methyl)phenyl)methanimine (7.5 mmol, 1.5 equiv.) and TBAI (0.25 mmol, 5 mol%). The reagents were dissolved in anhydrous DMA and the total volume of the solution was adjusted to 25 mL. The resulting mixture bubbled with an argon balloon for 20 min. After that, the reaction solution was introduced to the flow apparatus. The flow apparatus was purged with degassed argon to remove the air first. The syringe pump was then connected to the reaction mixture and 3.6 mL of polyfluoroalkoxy (PFA) microreactor coil (internal diameter of 1.0 mm) with a 34.5 kPa back-pressure regulator (BPR). The reaction was placed under 450 nm blue LEDs. The flow apparatus itself was set up with residence time (tR)=2 h, flow rate=1.8 mL/h. When the syringe was fully empty, a crude sample (1 mL) was taken from the collected solution and analyzed by determined through GC analysis. The yield of product 3 was determined to be 63%.
N-Cyclohexyl-1,1-bis(3-(trifluoromethyl)phenyl)meth-animine (3a):[16] 51.9 mg, 65% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.96 (s, 1H), 7.74 (d, J=7.9 Hz, 1H), 7.67~7.59 (m, 3H), 7.46~7.41 (m, 2H), 7.37 (d, J=7.6 Hz, 1H), 3.21~3.10 (m, 1H), 1.80~1.73 (m, 2H), 1.66~1.56 (m, 5H), 1.32~1.22 (m, 1H), 1.21~1.10 (m, 2H); 13C NMR (151 MHz, Chloroform-d) δ: 162.4, 140.3, 137.1, 131.7, 131.3, 131.0, 130.8, 129.3, 128.6, 126.5, 125.5, 124.7, 124.4, 124.1 (q, J=272.5 Hz), 123.9 (q, J=272.5 Hz), 61.9, 33.8, 25.5, 24.2; 19F NMR (564 MHz, Chloroform-d) δ: -63.0, -63.1.
N-tert-Pentyl-1,1-bis(3-(trifluoromethyl)phenyl)methan-imine (3b):[9] 64.3 mg, 83% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.94 (s, 1H), 7.71 (d, J=7.8 Hz, 1H), 7.64~7.53 (m, 3H), 7.48 (s, 1H), 7.40 (t, J=9.2 Hz, 2H), 1.61 (q, J=7.4 Hz, 2H), 1.03 (s, 6H), 0.97 (t, J=7.4 Hz, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 160.1, 142.0, 140.1, 131.6, 131.3, 130.7 (q, J=32.7 Hz), 130.6 (q, J=32.3 Hz), 128.7, 128.5, 126.2 (q, J=3.6 Hz), 125.2 (q, J=3.8 Hz), 125.0 (q, J=3.6 Hz), 124.4 (q, J=4.0 Hz), 124.1 (q, J=272.3 Hz), 123.8 (q, J=272.5 Hz), 60.0, 38.4, 28.3, 9.0; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.8.
N-tert-Butyl-1,1-bis(3-(trifluoromethyl)phenyl)methani-mine (3c):[9] 57.4 mg, 77% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.95 (s, 1H), 7.71 (d, J=7.9 Hz, 1H), 7.62~7.56 (m, 2H), 7.52 (d, J=7.9 Hz, 1H), 7.48 (s, 1H), 7.44~7.36 (m, 2H), 1.17 (s, 9H); 13C NMR (151 MHz, Chloroform-d) δ: 160.1, 141.9, 139.8, 131.7, 131.4, 130.8 (q, J=33.0 Hz), 130.6 (q, J=32.7 Hz), 128.7, 128.5, 126.2 (q, J=3.9 Hz), 125.2 (q, J=4.0 Hz), 125.1 (q, J=3.8 Hz), 124.4 (q, J=4.2 Hz), 124.1 (q, J=272.1 Hz), 123.8 (q, J=272.2 Hz), 57.6, 31.5; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.8.
N-(3-methyloxetan-3-yl)-1,1-bis(3-(trifluoromethyl)phe-nyl)methanimine (3d): 56.5 mg, 73% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.94 (s, 1H), 7.75 (d, J=7.9 Hz, 1H), 7.67 (d, J=8.2 Hz, 1H), 7.62 (t, J=7.8 Hz, 1H), 7.56 (d, J=7.9 Hz, 1H), 7.49~7.43 (m, 2H), 7.41 (d, J=7.7 Hz, 1H), 4.54 (d, J=5.7 Hz, 2H), 4.02 (d, J=6.4 Hz, 2H), 1.74 (s, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 162.5, 140.1, 137.7, 131.7, 131.3, 131.2 (q, J=33.1 Hz), 130.9 (q, J=32.6 Hz), 129.3, 128.8, 127.1 (q, J=3.7 Hz), 126.2 (q, J=4.0 Hz), 124.8 (q, J=3.5 Hz), 124.7 (q, J=3.8 Hz), 124.3 (q, J=272.5 Hz), 123.6 (q, J=272.7 Hz), 83.7, 62.2, 26.6; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.9. HRMS (ESI) calcd for C19H16F6NO [M+H]+ 388.1131, found 388.1129.
N-(Adamantan-1-yl)-1,1-bis(3-(trifluoromethyl)phenyl)-methanimine (3e):[9] 48.7 mg, 54% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.94 (s, 1H), 7.71 (d, J=7.9 Hz, 1H), 7.62~7.54 (m, 2H), 7.51 (d, J=7.9 Hz, 1H), 7.47 (s, 1H), 7.43~7.36 (m, 2H), 1.99 (s, 3H), 1.70 (s, 6H), 1.65~1.49 (m, 6H); 13C NMR (151 MHz, Chloroform-d) δ: 159.4, 142.0, 140.3, 131.5, 131.4, 130.6 (q, J=32.3 Hz), 130.5 (q, J=32.6 Hz), 128.6, 128.5, 126.2, 125.2, 124.9, 124.4, 124.0 (q, J=272.5 Hz), 123.8 (q, J=272.5 Hz), 58.7, 44.2, 36.2, 29.6; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.7.
N-(1-Methylcyclohexyl)-1,1-bis(3-(trifluoromethyl)-phenyl)methanimine (3f):[16] 56.2 mg, 68% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.95 (s, 1H), 7.71 (d, J=7.9 Hz, 1H), 7.64~7.55 (m, 3H), 7.49 (s, 1H), 7.45~7.38 (m, 2H), 1.69 (d, J=12.6 Hz, 2H), 1.61~1.53 (m, 3H), 1.50~1.43 (m, 2H), 1.31 (t, J=10.2 Hz, 3H), 1.02 (s, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 160.3, 142.0, 140.2, 131.3, 131.3, 130.7 (q, J=32.5 Hz). 130.6 (q, J=32.2 Hz), 128.7, 128.5, 126.2 (q, J=3.6 Hz), 125.2 (q, J=3.6 Hz), 124.7 (q, J=3.9 Hz), 124.3 (q, J=3.9 Hz), 124.0 (q, J=272.5 Hz), 123.8 (q, J=272.5 Hz), 59.3, 40.3, 28.1, 25.8, 22.8; 19F NMR (564 MHz, Chloroform-d) δ: -63.0, -63.1.
N-(4,4-Difluorocyclohexyl)-1,1-bis(3-(trifluoromethyl)-phenyl)methanimine (3g): 60.9 mg, 70% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.93 (s, 1H), 7.77 (d, J=7.9 Hz, 1H), 7.66 (q, J=7.9 Hz, 3H), 7.46 (t, J=7.8 Hz, 1H), 7.42 (s, 1H), 7.37 (d, J=7.5 Hz, 1H), 3.37~3.30 (m, 1H), 2.32~2.20 (m, 2H), 1.94~1.84 (m, 2H), 1.81~1.68 (m, 4H); 13C NMR (151 MHz, Chloroform-d) δ: 163.7, 139.8, 136.7, 131.7, 131.5 (q, J=33.0 Hz), 130.8 (q, J=32.9 Hz), 130.8, 129.6, 128.8, 126.9 (q, J=3.6 Hz), 125.8 (q, J=3.9 Hz), 124.7 (q, J=3.8 Hz), 124.1 (q, J=3.7 Hz), 124.0 (q, J=272.5 Hz), 123.9 (q, J=272.5 Hz), 57.9, 30.9 (t, J=24.5 Hz), 29.8 (t, J=4.9 Hz), 18.4; 19F NMR (564 MHz, Chloroform-d) δ: -65.3, -65.3, -99.2. HRMS (ESI) calcd for C21H18F8N [M+H]+ 436.1306, found 436.1306.
N-(Cyclohex-3-en-1-yl)-1,1-bis(3-(trifluoromethyl)phe-nyl)methanimine (3h): 48.4 mg, 61% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.97 (s, 1H), 7.74 (d, J=7.9 Hz, 1H), 7.63 (t, J=8.1 Hz, 3H), 7.50~7.42 (m, 2H), 7.38 (d, J=7.5 Hz, 1H), 5.71~5.60 (m, 2H), 3.47~3.33 (m, 1H), 2.40~2.27 (m, 1H), 2.17 (d, J=14.9 Hz, 1H), 2.03~1.82 (m, 3H), 1.74~1.65 (m, 1H); 13C NMR (151 MHz, Chloroform-d) δ: 163.2, 140.1, 137.0, 131.7, 131.3 (q, J=32.5 Hz), 131.0, 130.8 (q, J=32.4 Hz), 129.4, 128.7, 126.8, 126.6 (q, J=3.8 Hz), 125.6 (q, J=3.9 Hz), 124.8 (q, J=3.8 Hz), 124.6, 124.3 (q, J=4.0 Hz), 124.0 (q, J=272.6 Hz), 123.8 (q, J=272.6 Hz), 58.2, 32.4, 29.9, 23.7; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.7. HRMS (ESI) calcd for C21H17F6NNa [M+Na]+420.1157, found 420.1157.
N-(Cyclopent-3-en-1-yl)-1,1-bis(3-(trifluoromethyl)phe- nyl)methanimine (3i): 58.2 mg, 76% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.96 (s, 1H), 7.74 (d, J=7.9 Hz, 1H), 7.67~7.60 (m, 3H), 7.52~7.43 (m, 2H), 7.38 (d, J=7.6 Hz, 1H), 5.73 (s, 2H), 4.11~3.95 (m, 1H), 2.53 (d, J=6.4 Hz, 4H); 13C NMR (151 MHz, Chloroform-d) δ: 163.3, 140.1, 137.2, 131.8, 131.3, 131.3 (q, J=32.7 Hz), 130.8 (q, J=32.4 Hz), 129.3, 129.1, 128.7, 126.6 (q, J=4.0 Hz), 125.6 (q, J=3.8 Hz), 124.8 (q, J=3.8 Hz), 124.6 (q, J=3.6 Hz), 124.0 (q, J=272.5 Hz), 123.8 (q, J=272.5 Hz), 61.5, 41.4; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.8. HRMS (ESI) calcd for C20H16F6N [M+H]+ 384.1181, found 384.1200.
N-(Tetrahydrofuran-2-yl)-1,1-bis(3-(trifluoromethyl)-phenyl)methanimine (3j): 58.8 mg, 76% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 8.00 (s, 1H), 7.75 (d, J=7.9 Hz, 1H), 7.69~7.60 (m, 3H), 7.49~7.43 (m, 2H), 7.41 (d, J=7.7 Hz, 1H), 5.09 (dd, J=6.4, 3.7 Hz, 1H), 4.22 (q, J=7.4 Hz, 1H), 3.96~3.90 (m, 1H), 2.23~2.14 (m, 1H), 2.03~1.96 (m, 1H), 1.94~1.80 (m, 2H); 13C NMR (151 MHz, Chloroform-d) δ: 163.9, 139.5, 136.5, 132.1, 131.3, 131.3 (q, J=33.1 Hz), 130.9 (q, J=32.9 Hz), 129.4, 128.7, 127.2 (q, J=3.7 Hz), 126.0 (q, J=3.6 Hz), 125.2 (q, J=3.9 Hz), 124.7 (q, J=3.7 Hz), 124.0 (q, J=272.6 Hz), 123.8 (q, J=272.5 Hz), 91.5, 68.2, 34.1, 25.4; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.8. HRMS (ESI) calcd for C19H16F6NO [M+H]+ 388.1131, found 388.1125.
N-Cyclobutyl-1,1-bis(3-(trifluoromethyl)phenyl)metha-nimine (3k):[9] 36.4 mg, 49% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.97 (s, 1H), 7.74 (d, J=8.0 Hz, 1H), 7.66~7.61 (m, 3H), 7.45 (t, J=7.8 Hz, 1H), 7.38 (s, 1H), 7.32 (d, J=7.6 Hz, 1H), 3.96~3.89 (m, 1H), 2.35~2.25 (m, 2H), 2.12~2.06 (m, 2H), 1.92~1.85 (m, 1H), 1.76~1.65 (m, 1H); 13C NMR (151 MHz, Chloroform-d) δ: 163.4, 140.0, 137.3, 131.7, 131.2 (q, J=32.4 Hz), 131.1, 130.8 (q, J=32.7 Hz), 129.2, 128.7, 126.7 (q, J=3.8 Hz), 125.7 (q, J=3.7 Hz), 124.8 (q, J=4.0 Hz), 124.6 (q, J=4.0 Hz), 124.0 (q, J=272.4 Hz), 123.8 (q, J=272.6 Hz), 57.4, 31.3, 16.1; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.8.
N-Isopropyl-1,1-bis(3-(trifluoromethyl)phenyl)methani-mine (3l):[9] 39.5 mg, 55% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.96 (s, 1H), 7.73 (d, J=8.0 Hz, 1H), 7.66~7.59 (m, 3H), 7.46~7.41 (m, 2H), 7.37 (d, J=7.6 Hz, 1H), 3.52~3.45 (m, 1H), 1.2 (s, 3H), 1.2 (s, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 162.3, 140.2, 137.1, 131.7, 131.3 (q, J=32.7 Hz), 131.0, 130.8 (q, J=32.4 Hz), 129.4, 128.7, 126.6, 125.6, 124.8, 124.3 (q, J=3.8 Hz), 124.0 (q, J=272.7 Hz), 123.8 (q, J=272.5 Hz), 53.5, 23.8; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.8.
N-(1-Phenylpropan-2-yl)-1,1-bis(3-(trifluoromethyl)-phenyl)methanimine (3m): 58.3 mg, 67% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.91 (s, 1H), 7.63 (d, J=7.8 Hz, 2H), 7.55 (d, J=7.8 Hz, 1H), 7.49~7.38 (m, 2H), 7.22~7.15 (m, 3H), 7.00 (d, J=6.4 Hz, 2H), 6.76 (d, J=35.7 Hz, 2H), 3.61~3.42 (m, 1H), 2.99~2.91 (m, 1H), 2.87~2.79 (m, 1H), 1.30 (d, J=6.2 Hz, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 163.4, 139.9, 139.5, 137.0, 131.6, 131.0 (q, J=32.1 Hz), 130.7 (q, J=32.1 Hz), 130.8, 129.6, 129.0, 128.7, 128.2, 126.6 (q, J=4.1 Hz), 126.1, 125.3 (q, J=3.8 Hz), 124.7 (q, J=4.1 Hz), 124.1 (q, J=4.0 Hz), 124.0 (q, J=272.2 Hz), 123.7 (q, J=272.6 Hz), 60.3, 44.7, 22.2; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.6. HRMS (ESI) calcd for C24H20F6N [M+H]+ 436.1494, found 436.1494.
N-(Heptan-4-yl)-1,1-bis(3-(trifluoromethyl)phenyl)me-thanimine (3n): 46.5 mg, 56% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) 7.94 (s, 1H), 7.72 (d, J=8.7 Hz, 1H), 7.68~7.59 (m, 3H), 7.45 (t, J=7.8 Hz, 1H), 7.42 (s, 1H), 7.34 (d, J=7.6 Hz, 1H), 3.21 (dt, J=8.2, 4.6 Hz, 1H), 1.66~1.48 (m, 4H), 1.32~1.22 (m, 2H), 1.20~1.08 (m, 2H), 0.82 (t, J=7.3 Hz, 6H); 13C NMR (151 MHz, Chloroform-d) δ: 163.0, 140.2, 137.5, 131.6, 131.4, 131.1 (q, J=32.6 Hz), 130.8 (q, J=32.4 Hz), 129.2, 128.7, 126.5 (q, J=3.5 Hz), 125.3 (q, J=3.9 Hz), 124.8 (q, J=3.6 Hz), 124.7 (q, J=4.4 Hz), 124.0 (q, J=272.5 Hz), 123.9 (q, J=293.8 Hz), 62.2, 38.8, 19.7, 14.2; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.8. HRMS (ESI) calcd for C22H24F6N [M+H]+ 416.1807, found 416.1808.
N-Isobutyl-1,1-bis(3-(trifluoromethyl)phenyl)methani-mine (3o):[9] 26.9 mg, 36% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.95 (s, 1H), 7.74 (d, J=7.9 Hz, 1H), 7.69~7.61 (m, 3H), 7.46 (t, J=7.8 Hz, 1H), 7.41 (s, 1H), 7.36 (d, J=7.6 Hz, 1H), 3.18 (d, J=6.6 Hz, 2H), 2.11~2.02 (m, 1H), 0.94 (s, 3H), 0.93 (s, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 164.6, 140.0, 137.0, 131.6, 131.3, 131.3 (q, J=33.0 Hz), 130.8 (q, J=32.4 Hz), 129.4, 128.7, 126.7, 125.6, 124.7, 124.6, 124.0 (q, J=272.4 Hz), 123.7 (q, J=253.8 Hz), 61.8, 30.1, 20.7; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.7.
N-(Pent-4-en-1-yl)-1,1-bis(3-(trifluoromethyl)phenyl)-methanimine (3p): 23.9 mg, 31% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.95 (s, 1H), 7.74 (d, J=7.9 Hz, 1H), 7.68~7.61 (m, 3H), 7.46 (t, J=7.8 Hz, 1H), 7.42 (s, 1H), 7.36 (d, J=7.6 Hz, 1H), 5.88~5.69 (m, 1H), 5.25~4.62 (m, 2H), 3.37 (t, J=6.9 Hz, 2H), 2.12 (q, J=6.7 Hz, 2H), 1.85~1.77 (m, 2H); 13C NMR (151 MHz, Chloroform-d) δ: 164.9, 140.0, 138.2, 136.8, 131.6, 131.4 (q, J=32.6 Hz), 131.2, 130.9 (q, J=32.5 Hz), 129.4, 128.7, 126.7 (q, J=3.8 Hz), 125.7 (q, J=3.8 Hz), 124.7 (q, J=4.2 Hz), 124.5 (q, J=4.0 Hz), 124.0 (q, J=272.5 Hz), 123.7 (q, J=253.8 Hz), 114.8, 53.4, 31.5, 30.2; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.7. HRMS (ESI) calcd for C20H18F6N [M+H]+ 386.1338, found 386.1338.
tert-Butyl (3-(1,3-dioxoisoindolin-2-yl)propyl)carba-mate (3q): 32.2 mg, 53% yield, pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ: 7.83 (dd, J=5.4, 3.0 Hz, 2H), 7.71 (dd, J=5.5, 3.0 Hz, 2H), 5.06 (s, 1H), 3.75 (t, J=6.6 Hz, 2H), 3.14~3.10 (m, 2H), 1.87~1.80 (m, 2H), 1.40 (s, 9H); 13C NMR (151 MHz, Chloroform-d) δ: 168.6, 155.9, 134.1, 132.0, 123.3, 79.2, 37.4, 35.1, 28.8, 28.4. HRMS (ESI) calcd for C16H21N2O4 [M+H]+ 305.1496, found 305.1491.
N-(5-(2,5-Dimethylphenoxy)-2-methylpentan-2-yl)-1,1-bis(3-(trifluoromethyl)phenyl)methan-imine (3r):[9] 65.7 mg, 63% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.92 (s, 1H), 7.73 (d, J=7.8 Hz, 1H), 7.66~7.56 (m, 3H), 7.50 (s, 1H), 7.46~7.39 (m, 2H), 7.03 (d, J=7.4 Hz, 1H), 6.68 (d, J=7.5 Hz, 1H), 6.65 (s, 1H), 4.00 (t, J=6.3 Hz, 2H), 2.32 (s, 3H), 2.22 (s, 3H), 2.03~1.94 (m, 2H), 1.89~1.76 (m, 2H), 1.10 (s, 6H); 13C NMR (151 MHz, Chloroform-d) δ: 160.4, 157.1, 141.8, 139.9, 136.5, 131.6, 131.3, 130.8 (q, J=32.8 Hz), 130.6 (q, J=32.3 Hz), 130.3, 128.8, 128.6, 126.3 (q, J=3.6 Hz), 125.3 (q, J=3.7 Hz), 125.0 (q, J=3.9 Hz), 124.4 (q, J=4.0 Hz), 124.1 (q, J=272.6 Hz), 123.8 (q, J=272.7 Hz), 123.6, 120.7, 112.1, 68.4, 59.6, 42.8, 28.8, 25.0, 21.4, 15.8; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.7.
N-((1R,4aR,4bR,10aR)-7-Isopropyl-1,4a-dimethyl-1,2,3,4,4a,4b,5,6,10,10a-decahydrophenanthren-1-yl)-1,1-bis-(3-(trifluoromethyl)phenyl)methanimine (3s): 73.4 mg, 64% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.78 (s, 1H), 7.69 (d, J=7.9 Hz, 1H), 7.57 (q, J=7.8 Hz, 3H), 7.44 (s, 1H), 7.41~7.36 (m, 2H), 5.84 (s, 1H), 5.53 (s, 1H), 2.51 (d, J=18.8 Hz, 1H), 2.28~2.20 (m, 1H), 2.10 (d, J=10.8 Hz, 2H), 2.06 (s, 1H), 1.99 (d,J=13.0 Hz, 1H), 1.80 (d, J=12.0 Hz, 2H), 1.52~1.37 (m, 4H), 1.30~1.20 (m, 2H), 1.14~1.07 (m, 1H), 1.03 (t, J=6.5 Hz, 6H), 0.97 (s, 3H), 0.82 (s, 3H); 13C NMR (151 MHz, Chloroform-d) δ: 159.0, 145.2, 142.3, 140.3, 135.4, 131.7, 131.2, 130.6 (q, J=32.4 Hz), 130.5 (q, J=32.5 Hz), 128.6, 128.4, 126.1 (q, J=3.8 Hz), 125.1 (q, J=4.0 Hz), 125.0 (q, J=4.7 Hz), 124.3 (q, J=4.0 Hz), 124.0 (q, J=272.4 Hz), 123.8 (q, J=272.7 Hz), 122.5, 121.8, 62.4, 52.9, 51.3, 40.4, 38.1, 35.5, 34.9, 27.6, 24.7, 22.7, 22.0, 21.4, 20.9, 19.4, 13.8; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -62.7. HRMS (ESI) calcd for C34H38F6N [M+H]+574.2903, found 574.2896.
N-(2-(4-Chlorophenoxy)propan-2-yl)-1,1-bis(3-(trifluo-romethyl)phenyl)methanimine (3t): 59.2 mg, 61% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.87 (s, 1H), 7.63 (d, J=7.7 Hz, 1H), 7.56 (dd, J=5.5, 3.0 Hz, 2H), 7.51~7.48 (m, 3H), 7.41 (t, J=7.8 Hz, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.25 (s, 1H), 7.16 (t, J=7.8 Hz, 1H), 7.09 (d, J=7.9 Hz, 1H), 2.11 (s, 6H); 13C NMR (151 MHz, Chloroform-d) δ: 168.3, 159.5, 141.3, 137.1, 134.4, 133.7, 131.8, 131.1, 130.6 (q, J=32.5 Hz), 130.3 (q, J=33.0 Hz), 130.8, 128.6 (q, J=3.2 Hz), 126.8 (q, J=3.7 Hz), 125.1 (q, J=3.9 Hz), 124.8 (q, J=3.8 Hz), 124.0 (q, J=272.4 Hz), 123.9 (q, J=4.1 Hz), 123.9 (q, J=272.3 Hz), 122.4, 76.4, 30.4; 19F NMR (564 MHz, Chloroform-d) δ: -62.6, -63.0. HRMS (ESI) calcd for C24H19ClF6NO [M+H]+ 486.1054, found 486.1053.
N-(But-3-en-1-yl)-1,1-bis(3-(trifluoromethyl)phenyl)-methanimine (3u):[9] 14.8 mg, 20% yield, colorless oil. 1H NMR (600 MHz, Chloroform-d) δ: 7.95 (s, 1H), 7.74 (d, J=7.9 Hz, 1H), 7.68~7.61 (m, 3H), 7.45 (t, J=7.8 Hz, 1H), 7.43 (s, 1H), 7.36 (d, J=7.6 Hz, 1H), 5.89~5.73 (m, 1H), 5.10~4.97 (m, 2H), 3.44 (t, J=7.0 Hz, 2H), 2.47 (q, J=7.4, 6.9 Hz, 2H); 13C NMR (151 MHz, Chloroform-d) δ: 165.1, 139.9, 136.7, 136.3, 131.6, 131.4 (q, J=32.7 Hz), 130.9 (q, J=32.6 Hz), 131.2, 129.4, 128.7, 126.8 (q, J=3.9 Hz), 125.7 (q, J=4.0 Hz), 124.7 (q, J=4.0 Hz), 124.6 (q, J=3.8 Hz), 124.0 (q, J=272.4 Hz), 123.8 (q, J=272.7 Hz), 116.2, 53.7, 35.4; 19F NMR (564 MHz, Chloroform-d) δ: -62.7, -62.7.
Supporting Information 1H NMR and 13C NMR spectra of compounds 3a~3u, and the radical trapping experiments. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(Lu, Y.)