


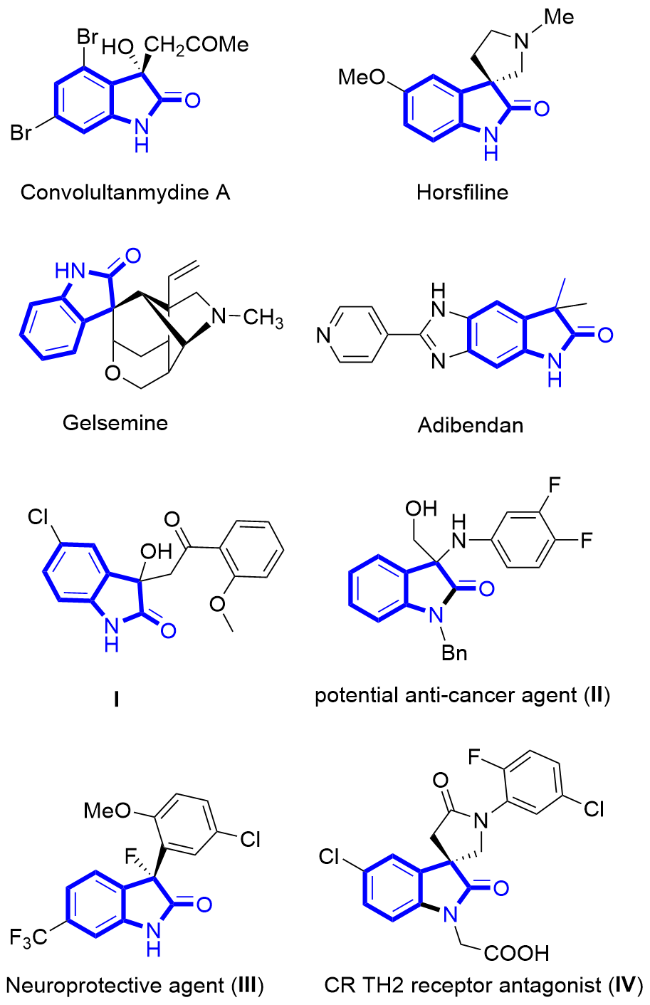
吲哚酮类化合物是以吲哚为母体而衍生出的一类杂环化合物, 它们广泛地存在于哺乳动物的体液和组织, 具有重要的生理作用[1-19]. 吲哚酮骨架也广泛地存在于具有生物活性的天然产物和药物分子中(图1). 例如, Convolultanmydine A[20]是一种从海洋苔藓虫分离出的生物碱, 它对人体内的白血病细胞的分化有较强的抑制作用; Horsfiline[21]是从植物中分离出的生物碱, 它拥有止痛的效果. Gelsemine[22]是存在于断肠草中的生物碱, 它有剧毒; Adibendan[23]是一种对抗心力衰竭的药物; 化合物I[3]具有对抗艾滋病毒的潜力; 化合物II[24]是一种有潜力的抗癌药物; 化合物III[25]是一种神经保护药物, 它具有开启钾离子通道的作用; 化合物IV[26]是一种CR TH2受体拮抗剂. 2-吲哚酮是各种吲哚酮衍生物的核心, 通过使用不同的取代基在其3号位进行修饰, 可以得到许多种吲哚酮衍生物, 它们展现出不同的生物活性, 如抗病毒、抗微生物及抗风湿等[3]. 鉴于吲哚酮化合物广泛的生物活性和潜在的药用价值, 因此, 发展高效的合成方法构建吲哚酮化合物具有重要的意义.
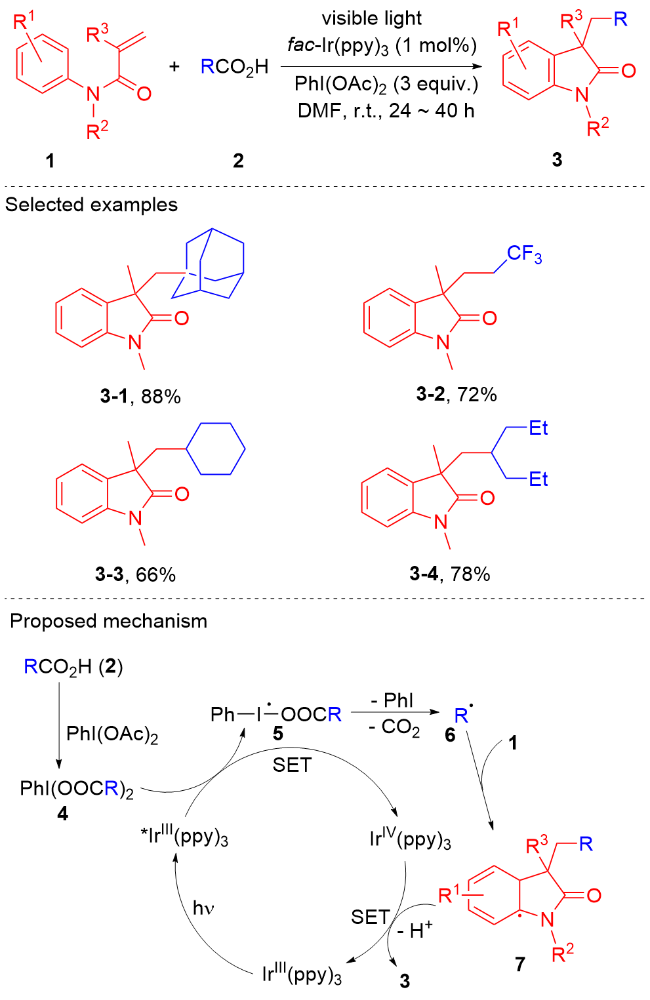
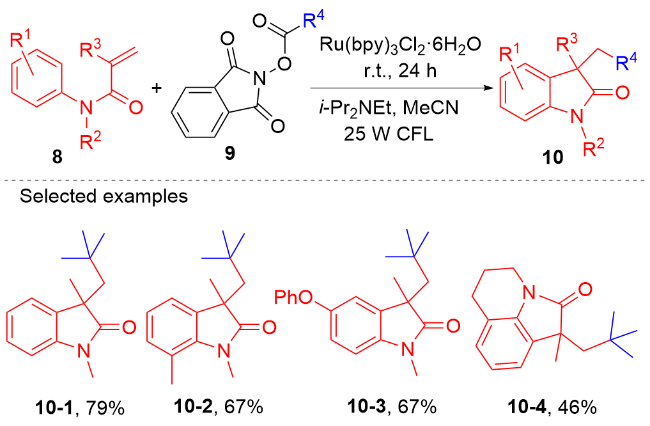
在过去的几十年, 科学家发展了大量的方法用于合成各种各样的吲哚酮衍生物[26-46]. 吲哚酮衍生物的最直接的合成方法是通过预先存在的吲哚酮骨架的功能化反应[29-32]. 此外, 过渡金属催化的反应也能用于吲哚酮衍生物的合成[33-36]. 此类反应通常要求高温、昂贵的过渡金属和配体等苛刻的条件, 而且这类反应的底物通常需要预先官能团化, 限制了反应的底物范围. 在过去的十年, 通过自由基引发的N-芳基丙烯酰胺加成环化反应[37-42]合成吲哚酮骨架的策略吸引了有机合成界的关注. 这种策略尽管取得了重要研究进展, 但是受限于高温和有毒的自由基引发剂. 为了克服这些缺陷, 光氧化还原催化的N-芳基丙烯酰胺的自由基加成环化串联反应[26-28]以及氢芳化反应[43-46]成为了合成吲哚酮衍生物的一种强有力的策略. 本文将根据自由基类型综述2013年以来光氧化还原催化的N-芳基丙烯酰胺自由基加成环化反应及氢芳基化反应取得的研究进展.
1 N-芳基丙烯酰胺参与的自由基串联反应
1.1 烷基自由基
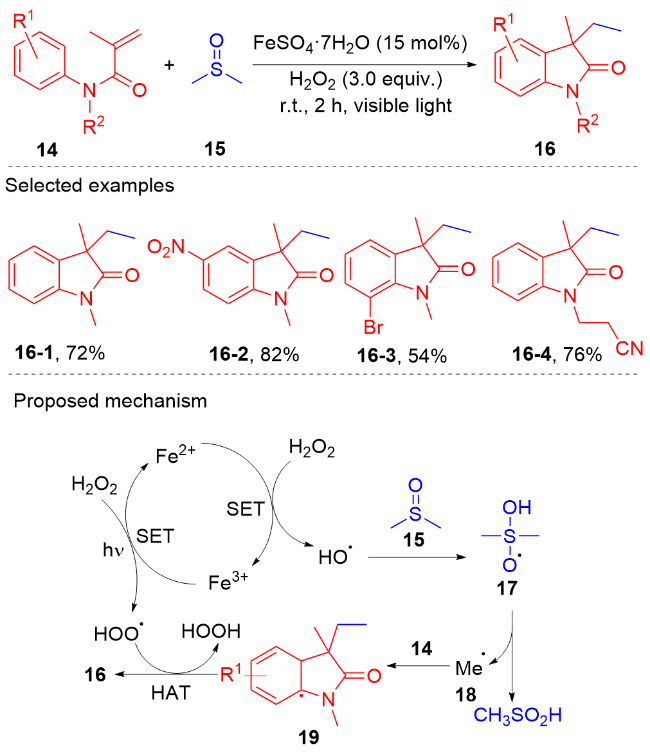
2017年, 王磊等[51]发展了一种可见光诱导、铁催化的N-芳基丙烯酰胺的甲基芳化反应(Scheme 4). 此反应使用二甲基亚砜作为甲基源, 催化量的二价铁盐为催化剂, 3 equiv.的双氧水为氧化剂, 在可见光的辐射下反应2 h, 以中等到良好的产率获得了各种甲基化的吲哚酮产物. 根据他们提出的机理, 催化量的二价铁盐将双氧水分解成羟基自由基, 进一步与二甲基亚砜发生亲电加成反应获得中间体17, 其脱去甲基亚磺酸生成甲基自由基. 在双氧水的作用下, 甲基自由基与N-芳基丙烯酰胺反应得到甲基化的吲哚酮衍生物. 这种策略最大的优势是不需要使用光敏剂. 遗憾的是, 此策略只限于二甲基亚砜, 其它的砜类如甲基苯基亚砜、正丙基亚砜和正丁基亚砜在该体系下不能反应.
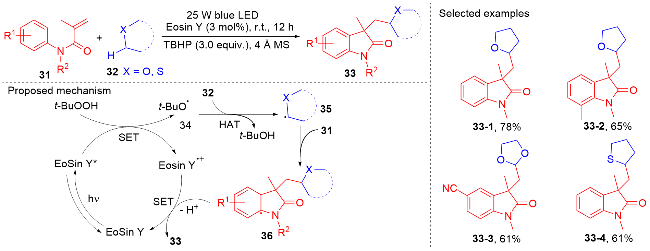
2019年, 王磊课题组[54]报道了曙红Y (Eosin Y)/叔丁基过氧化氢(TBHP)协同作用的光催化体系, 实现了简单醚与N-芳基丙烯酰胺的自由基加成环化反应(Scheme 7). 在该反应中, 醚既为底物也为溶剂, 4 Å分子筛作添加剂, 以49%~83%的产率获得了一系列含有醚的吲哚酮化合物. 不论苯环上含吸电子取代基还是给电子取代基的N-芳基丙烯酰胺的在该体系下都能兼容, 苯环上取代基的位置对反应产率的影响不大; 醚的范围有四氢呋喃、四氢噻吩、乙醚及1,3-二氧戊烷等简单醚. TBHP既作自由基引发剂也作氢原子转移剂. 作者提出TBHP被激发态的光敏剂Eosin Y*还原形成叔丁氧自由基, 进而攫取醚邻位的氢原子生成烷基自由基.
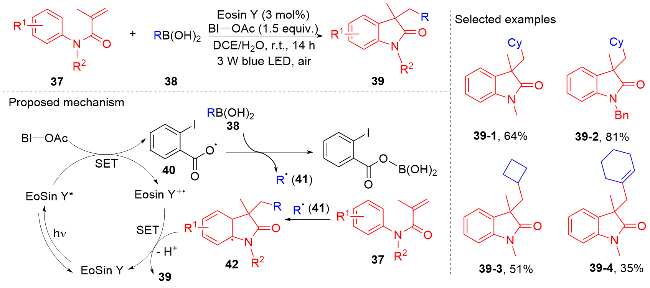
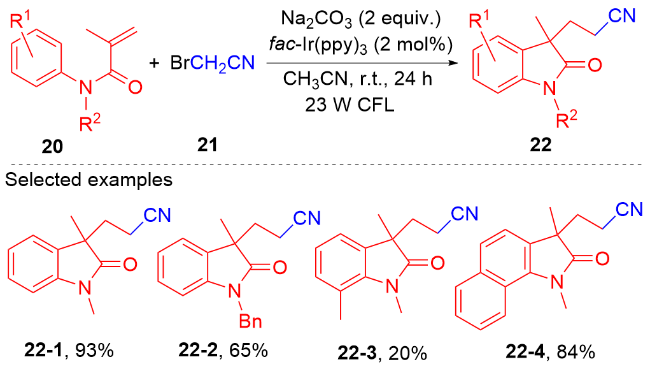
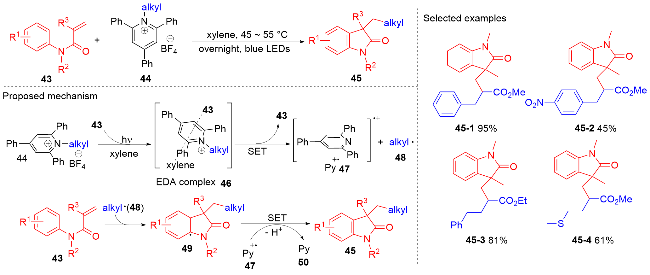
烷基伯胺是一种丰富的天然原料, 它能被用于制备Katritzky类型吡啶盐. 近些年, Katritzky盐的脱氨化过程被证明是一种产生烷基自由基的新方法[56]. 2021年, 陈祥雨课题组[57]利用Katritzky盐作烷基自由基前体, 实现了烷基吲哚酮骨架的合成(Scheme 9). 该反应使用二甲苯作为溶剂, 蓝灯作为光源, 无需使用光催化剂和添加剂, 对各种各样的N-芳基丙烯酰胺都能兼容. 研究表明Katritzky盐和N-芳基丙烯酰胺在蓝光照射下能形成电子供体-受体(EDA)复合物, 该复合物分解产生烷基自由基和三苯基吡啶自由基正离子. 在三苯基吡啶自由基正离子的作用下, 烷基自由基和N-芳基丙烯酰胺进行自由基加成环化反应生成烷基吲哚酮产物. 密度泛函理论(DFT)研究表明, 二甲苯溶剂分子能显著提高与Katritzky盐形成的EDA复合物的光活性.
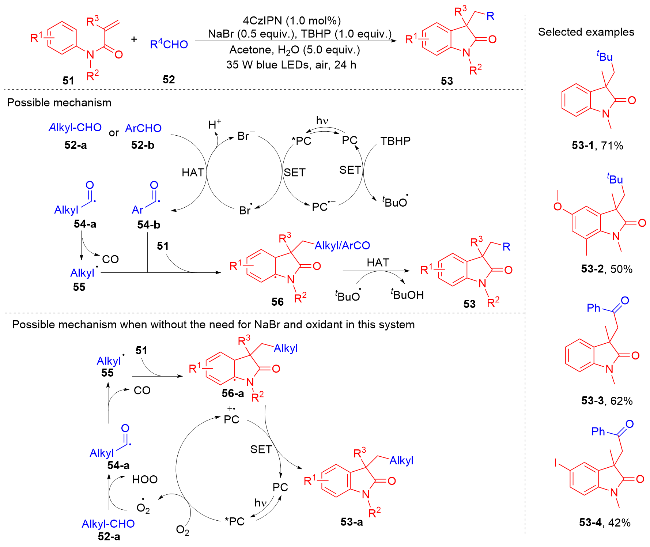
2022年, 黄华文等[58]报道了光氧化还原介导的醛与N-芳基丙烯酰胺的自由基加成环化反应(Scheme 10). 以4CzIPN为光催化剂, 溴化钠与水为添加剂, 叔丁基过氧化氢为氧化剂, 丙酮为溶剂, 在蓝光的辐射下以中等到良好的产率获得了一系列烷基化的吲哚酮衍生物和芳酰化的吲哚酮衍生物. 当使用脂肪醛时, 在光敏剂4CzIPN和溴化锂的共同作用下, 烷基醛被氧化为不稳定的酰基自由基中间体54-a, 它脱羰生成烷基自由基; 当醛为芳基醛时, 它被氧化为稳定的芳酰基自由基. 叔丁基过氧化氢在此体系下被还原为叔丁氧自由基, 在叔丁基氧自由基的作用下, 烷基自由基和芳酰基自由基与N-芳基丙烯酰胺进行自由基加成环化反应分别得到烷基化的吲哚酮产物和芳酰化的吲哚酮产物. 值得注意的是, 在溴化钠和叔丁基过氧化氢不存在的情况下, 激发态的4CzIPN被空气中的氧气氧化淬灭, 得到的氧气自由基负离子能氧化烷基醛形成中间体54-a.
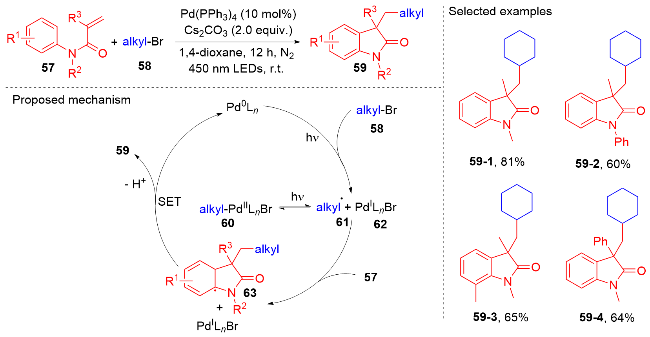
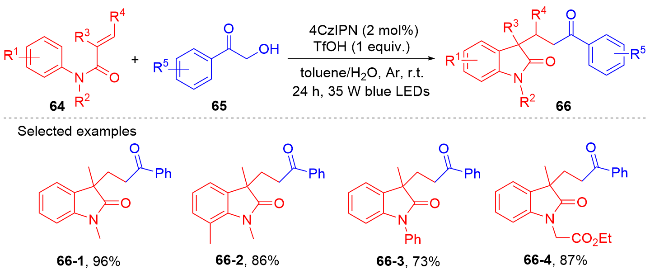
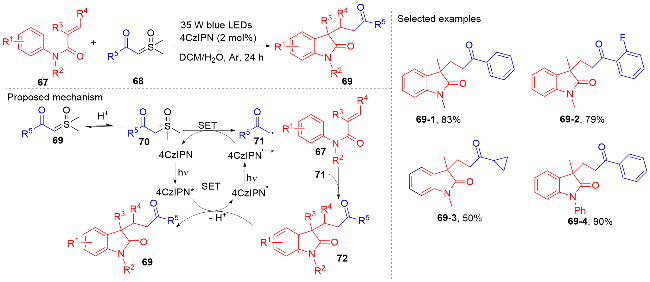
1.2 酰基自由基
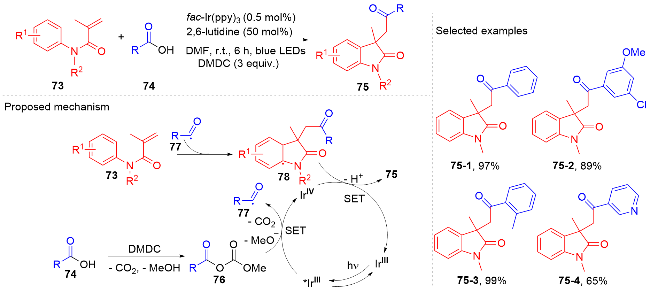
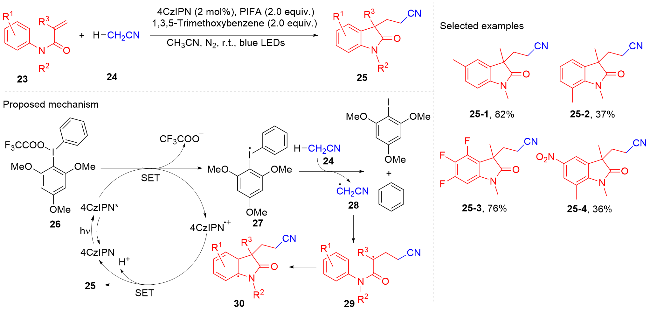
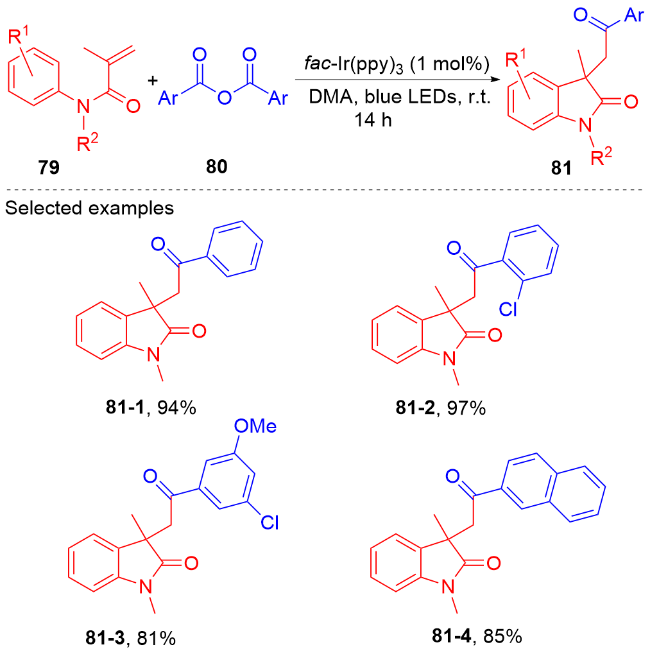
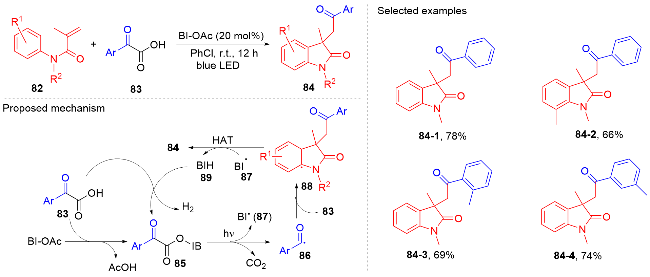
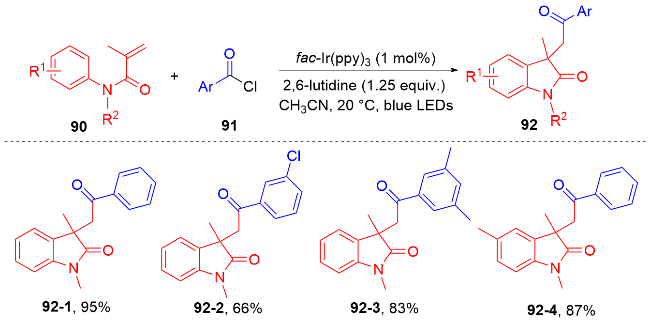
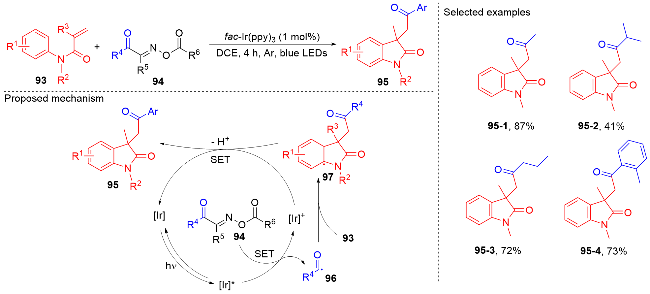
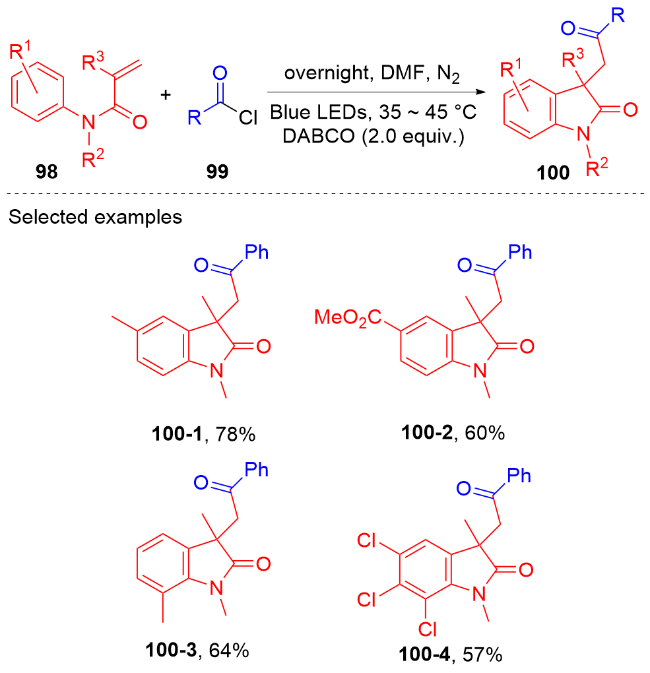
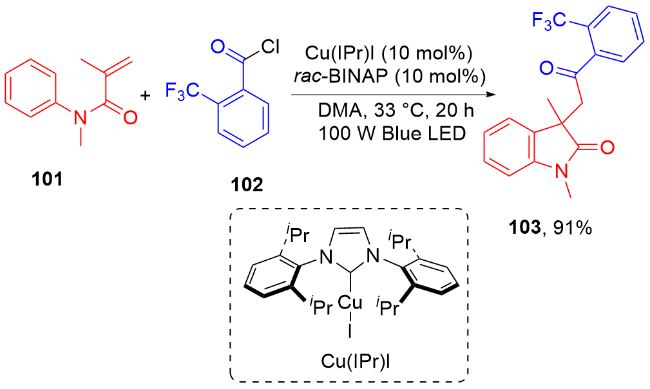
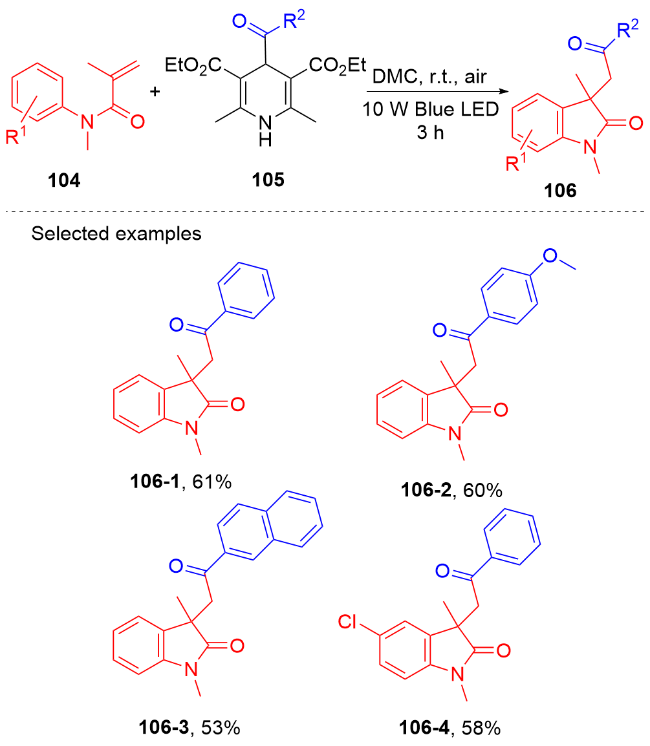
羧酸与二甲基二碳酸酯(DMDC)原位形成相应的酸酐. 在光敏剂fac-Ir(ppy)3的作用下, 酸酐脱去二氧化碳和甲醇负离子形成芳酰基自由基, 它与N-芳基丙烯酰胺进行自由基加成环化反应得到酰化的吲哚酮骨架. 该方法展现宽广的底物范围和优秀的官能团兼容度, 遗憾的是, 该反应只限芳基羧酸, 脂肪羧酸在该体系下不能反应.
1.3 含氟烷基自由基
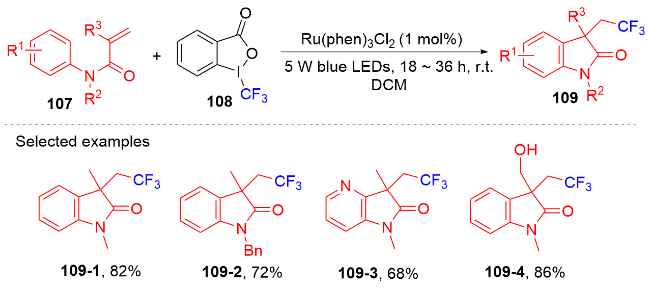
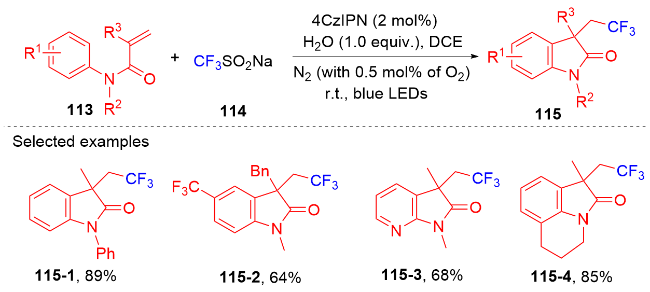
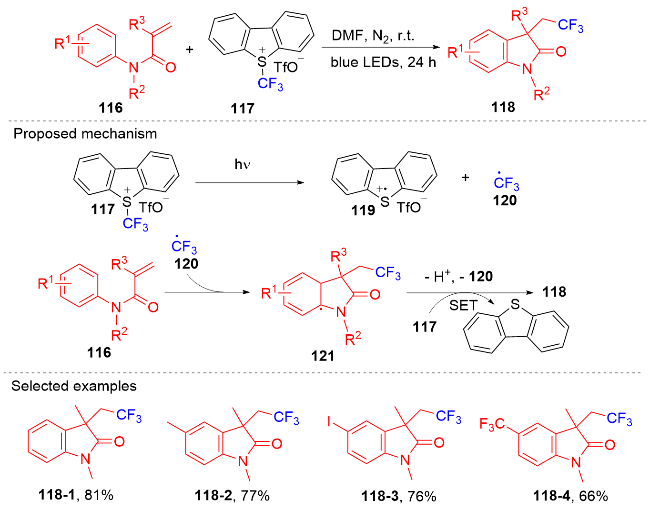
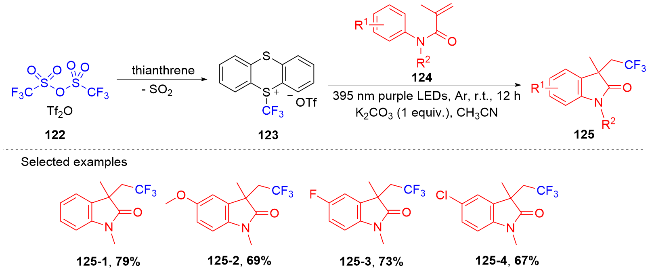
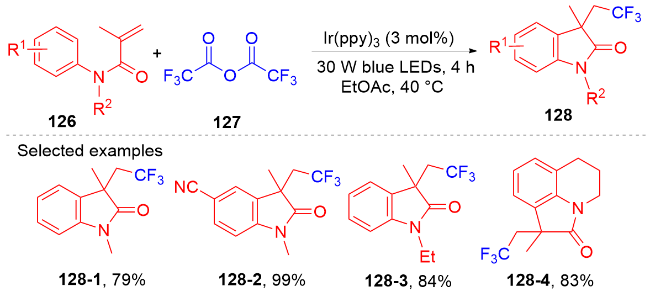
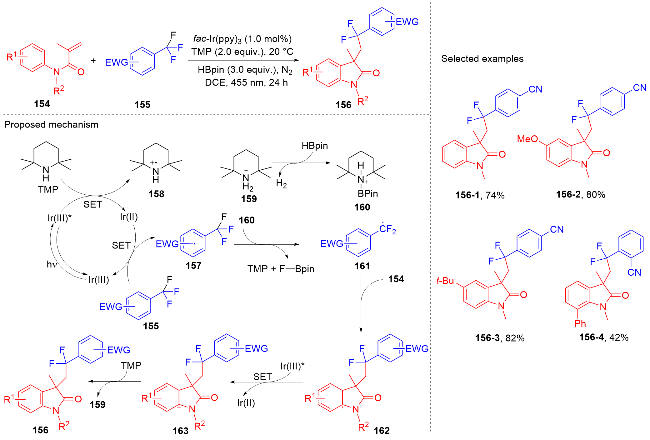
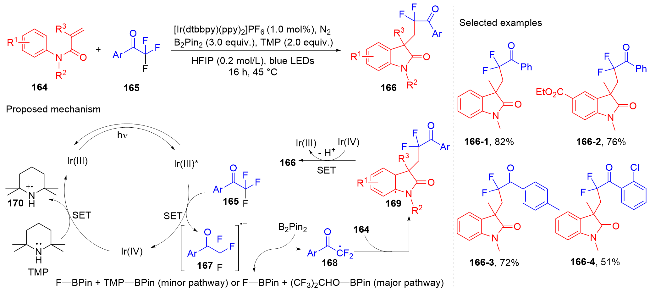
芳基二氟甲基化合物是药物化学中重要的结构单元, 将简单易得的三氟甲基芳烃转化为二氟甲基化合物具有高度的原子和步骤经济性. 然而, 三氟甲基芳烃的碳氟键断裂是非常有挑战性的, 因为C(sp3)—F键非常惰性, 所以选择性单个碳氟键断裂非常困难. 2017年, König课题组[80]报道了光催化和路易斯酸活化相结合的三氟甲基选择性单个碳氟键断裂的策略(Scheme 32). 此策略能应用于含氟吲哚酮骨架的构建. 根据作者提出的机理, 质子化的四甲基哌啶与频哪醇硼烷脱去氢气形成中间体160. 在fac-Ir(ppy)3的作用下三氟甲苯衍生物被还原成中间体157, 后者在中间体160的作用下脱掉氟负离子得到二氟苄基自由基161; 其与N-芳基丙烯酰胺反应得到二氟甲基化的吲哚酮产物. 值得注意的是, 三氟甲基底物必须另外带吸电子基团才能使碳氟键断裂.
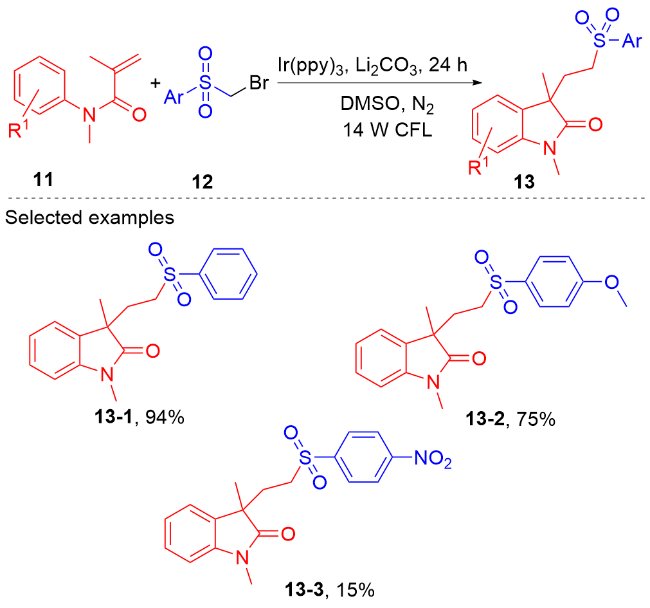
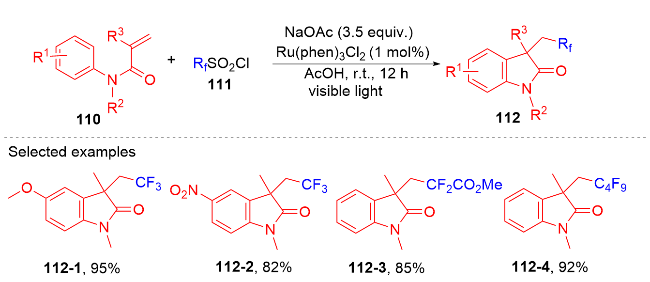
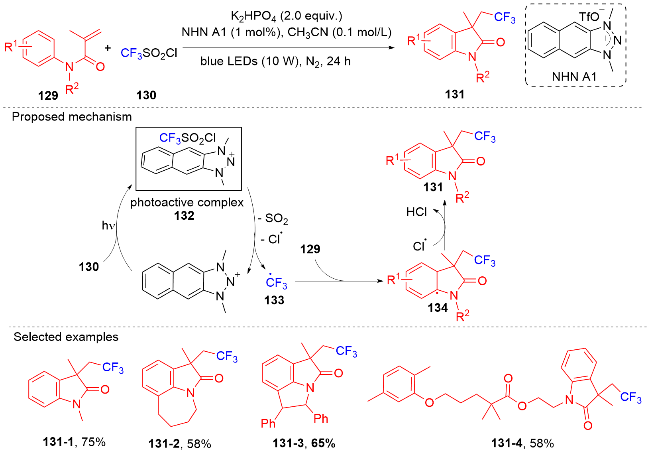
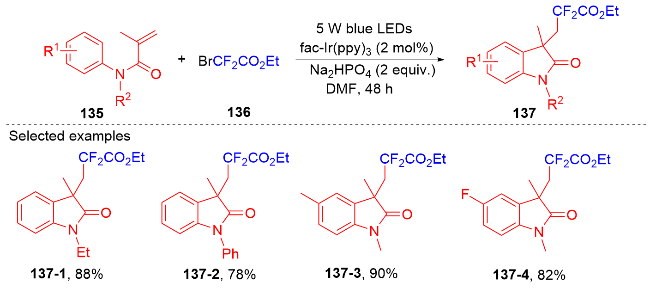
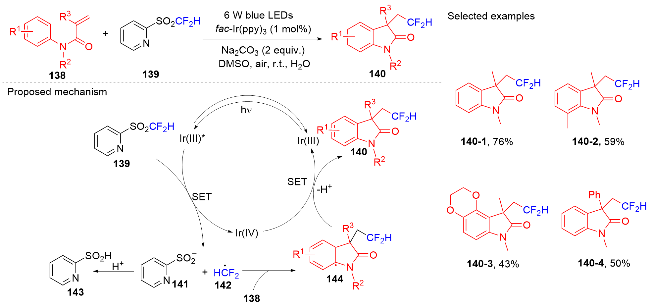
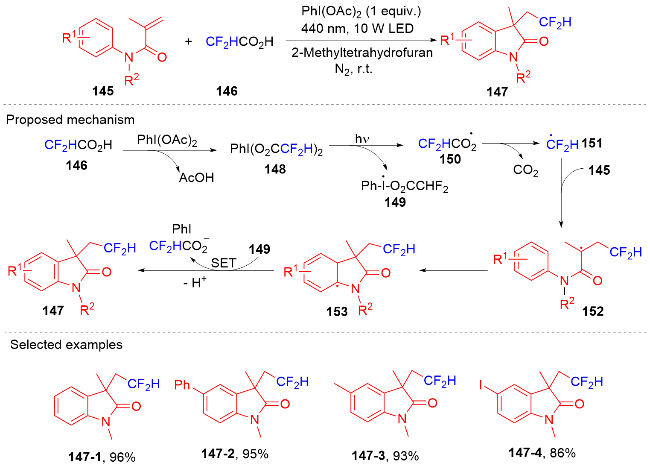
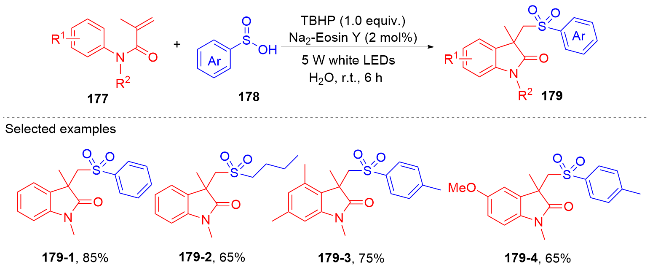
1.4 磺酰基自由基参与吲哚酮骨架的构建
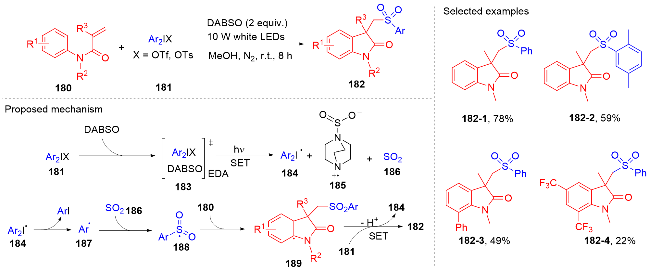
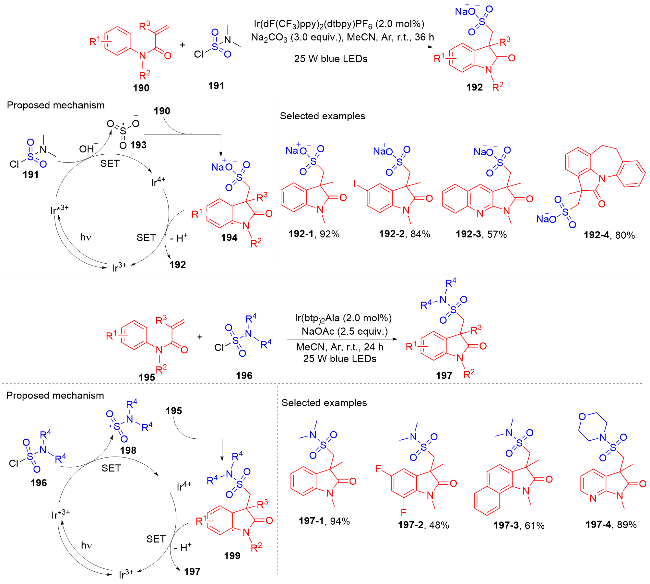
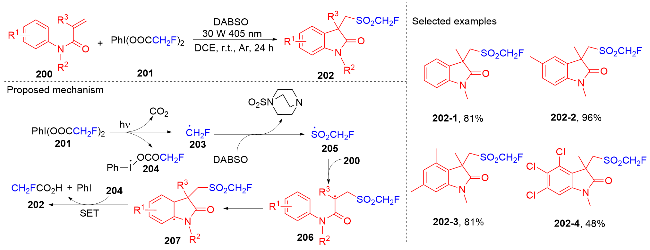
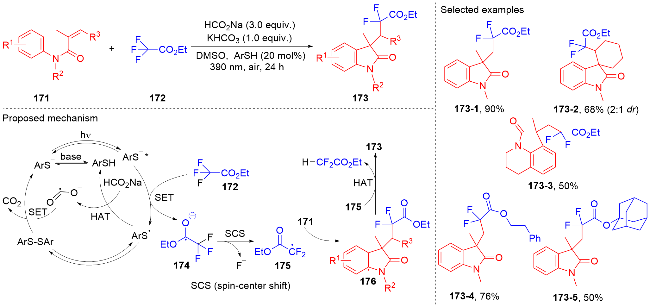
1.5 其它类型自由基
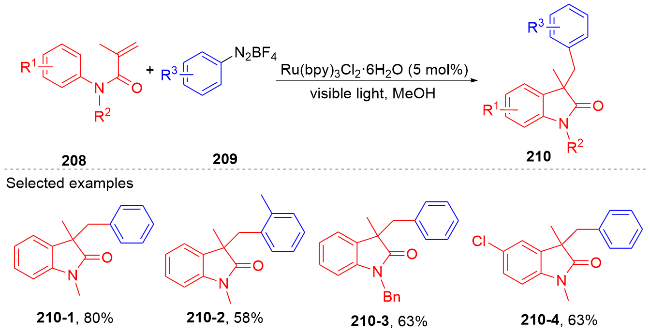
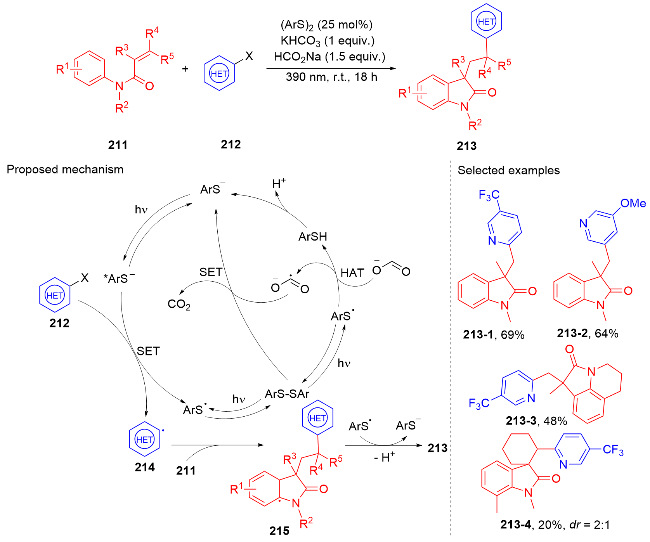
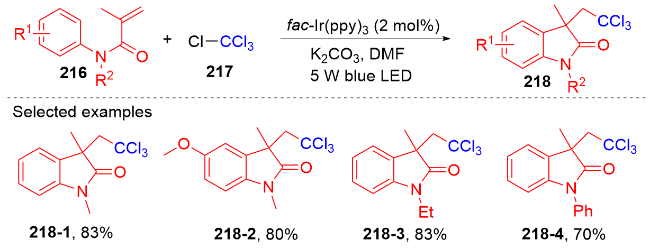
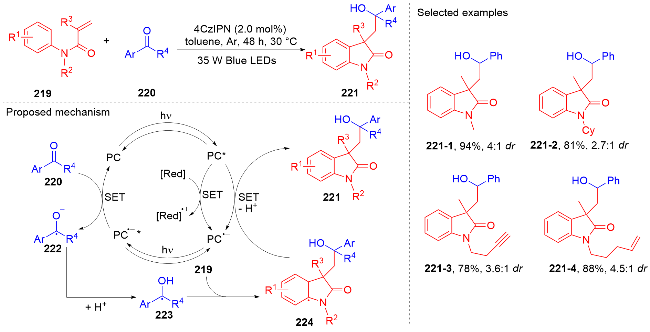
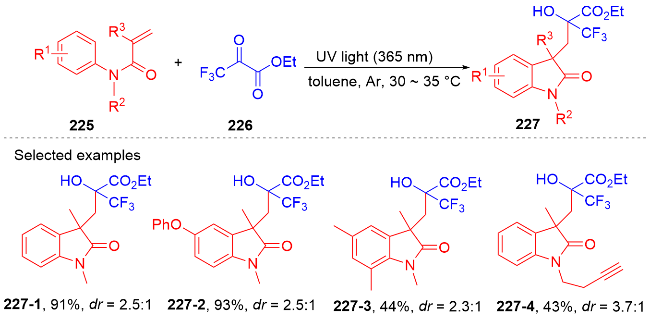
2 N-芳基丙烯酰胺的氢芳化反应
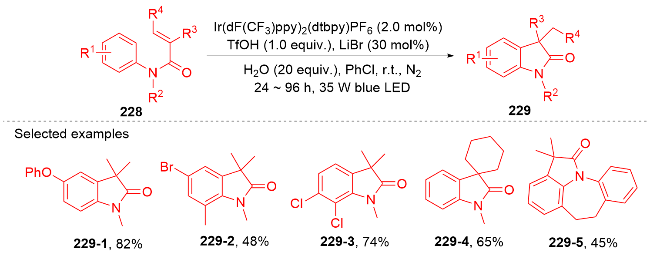
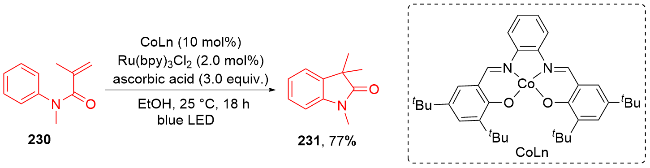
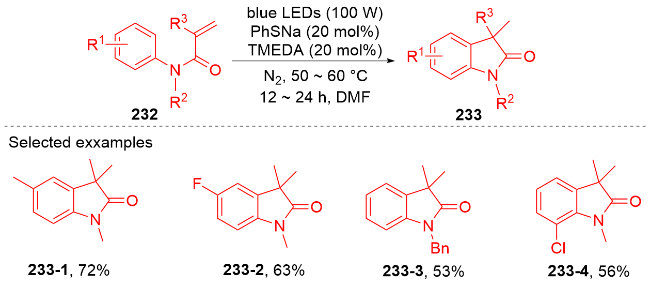
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 总结与展望
本文综述了近年来光介导的N-芳基丙烯酰胺合成吲哚酮衍生物的反应, 主要包括N-芳基丙烯酰胺与烷基自由基前体、含氟烷基自由基前体、酰基自由基前体、磺酰基自由基前体以及其它类型自由基前体的自由基串联反应及其氢芳化反应. 光介导的N-芳基丙烯酰胺参与的自由基串联反应及其氢芳化反应发展迅速, 催化体系越来越简单, 自由基前体越来越丰富, 构筑的吲哚酮衍生物范围越来越广泛. 它们已经成为构筑吲哚酮衍生物的重要策略. 尽管N-芳基丙烯酰胺参与的自由基串联反应及氢芳化反应已经取得了重要进展, 但是, 目前仍然存在一些有待解决的问题: (1)大部分光催化的自由基串联反应需要使用昂贵的过渡金属催化剂, 其成本问题和金属残留问题将不可避免地影响其深入拓展和应用; (2)在已报道的光介导的N-芳基丙烯酰胺参与的自由基串联反应中, 通常需要使用光催化剂和添加剂, 无催化剂光催化反应有待进一步发展. 随着近年光、电和机械化学等环境友好合成策略的发展, N-芳基丙烯酰胺的自由基型环合反应必将取得进一步研究进展.
(Zhao, C.)