


可见光催化反应利用清洁、绿色的可再生太阳能作为驱动力, 具有反应条件温和、反应速度快、环境友好等优点, 逐渐成为化学合成中的一种重要手段[1]. 通过光催化可以在温和的反应条件下生成多种高活性的自由基中间体, 从而实现许多在传统热反应条件下无法实现的化学转化. 然而, 自由基中间体的高反应性带来了化学选择性和对映选择性控制的巨大挑战[2-4]. 随着可见光催化技术的不断发展, 化学家们对反应选择性, 尤其是对映选择性的要求越来越高. 同时, 可持续发展理念在化学合成中的推进促使化学家们不断寻找新的催化剂, 以更高效和环保的方式实现化合物的对映选择性合成. 酶作为自然界中的天然催化剂, 具有温和、绿色的反应条件, 其活性口袋内独特的空间结构能够很好地控制反应的对映选择性. 此外, 底物在酶的活性位点内进行反应, 也能够有效地控制化学选择性[5]. 因此, 科学家们开始探索将光催化与酶催化相结合, 即光酶催化, 以利用酶的高选择性来提升自由基反应的选择性.
在诸多光酶催化的不对称自由基反应中, 自由基加成反应在近几年发展迅速, 展现了其在有机合成中的广阔应用前景[6-8]. 鉴于目前缺乏系统性的总结, 我们认为有必要对这些成果进行全面报道, 综述其反应机理和优势, 并讨论其中存在的问题. 期望填补相关研究领域的空白, 为研究人员提供有价值的参考, 推动光酶催化策略在不对称自由基加成反应中的进一步发展, 支持构建更加丰富的反应类型. 根据反应类型, 这些自由基加成反应可以分为分子内自由基加成和分子间自由基加成两大类. 同时, 按照自由基的生成方式, 每一种反应类型又进一步划分为单电子还原途径和单电子氧化途径. 本文将对这些反应的最新研究进展进行系统总结与分析, 探讨其反应机理、优势及存在的问题, 并展望未来的发展方向. 通过对光酶催化策略在不对称自由基加成反应中的应用进行全面综述, 我们期望为研究人员提供有价值的参考, 推动这一领域的进一步发展.
1 分子内自由基加成
1.1 单电子还原途径
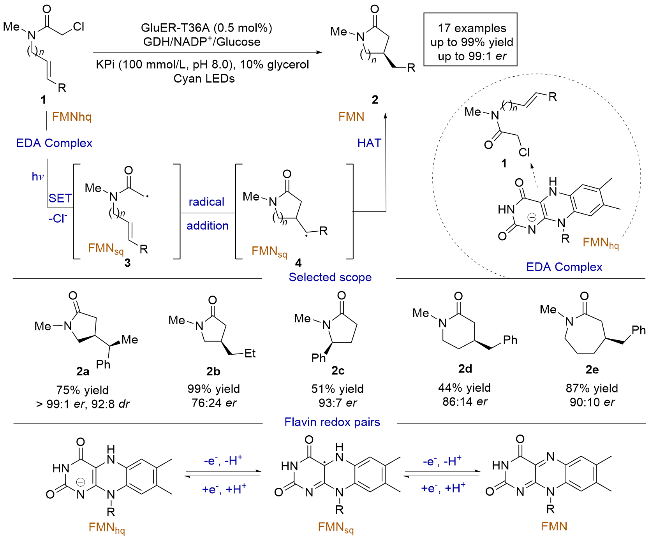
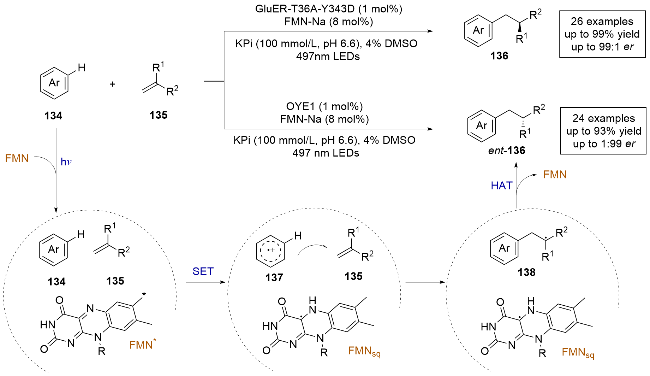
2019年, Hyster课题组[9]报道了一例光酶催化α-氯代酰胺与非活化烯烃发生的分子内不对称自由基环加成反应(Scheme 1). 传统方法中, 亲电自由基和非活化烯烃的不对称自由基加成存在区域选择性的问题, 并且缺乏能够向加成后生成的前手性自由基中间体不对称给氢的试剂. 本研究巧妙地解决了这些难题, 以优异的产率和对映选择性得到了目标产物. 研究团队采用了黄素依赖的烯烃还原酶(flavin-dependent “ene”-reductases, EREDs). 机理研究表明, 原位生成的黄素-对苯二酚(FMNhq)与α-氯代酰胺生成的电子供体-受体(electron donor-acceptor complex, EDA)复合物[10-11]在光照条件下直接激发, α-氯代酰胺1还原脱氯后生成自由基中间体3, 随后与烯烃发生分子内的自由基加成, 生成手性碳自由基中间体4, 最后在酶的引导下与黄素半醌(FMNsq)发生氢原子转移(Hydrogen atom transfer, HAT). 通过这种策略, 可以得到对映体富集的五元、六元、七元等环化产物2. 当使用三取代烯烃时, 该方法也能很好地控制产物的对映选择性和非对映选择性, 表明酶可以精确控制碳-碳键形成和HAT步骤的立体选择性. 该工作证明了可见光可以激发酶口袋中底物和辅助因子生成的络合物, 为开发天然酶的非天然转化提供了新的思路.
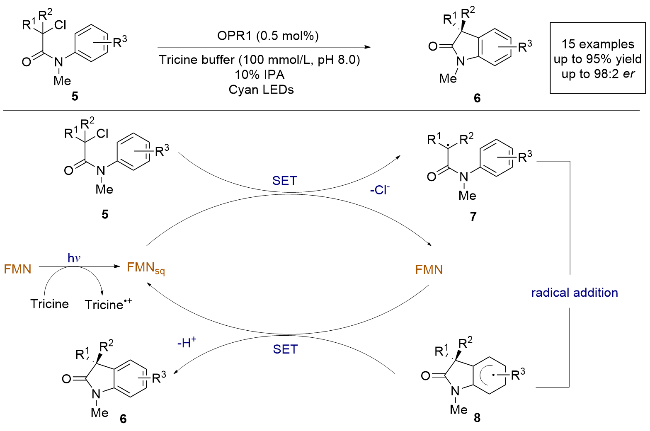
依赖黄素的“烯”还原酶(EREDs)是一类用于实现立体选择性还原的催化剂, 通常通过氢化物转移机制进行. 然而, 2019年, Hyster课题组[12]发现EREDs还可以通过单电子转移机制催化还原脱卤和环化反应, 使α-卤代酰胺生成具有对映选择性的氧化吲哚(Scheme 2), 这种C—C键形成反应在自然界中尚未被发现, 目前也没有相关的催化不对称实例. 研究中采用了黄素依赖的烯烃还原酶(OPR1), 无需外加辅助因子循环系统, 并使用三(羟甲基)甲基甘氨酸缓冲体系(tricine buffer)作为还原剂. 其反应机理如下: 在光照条件下, 黄素单核苷酸(FMN)被tricine还原为FMNsq, 随后底物5被FMNsq单电子还原, 伴随着氯负离子的离去, 生成碳中心自由基中间体7, 该自由基中间体7对芳环进行不对称自由基加成, 然后再被FMN氧化芳构化, 最终生成环化产物6. 这项研究不仅展示了黄素依赖酶在光催化自由基反应中的潜力, 还表明了无需外加循环系统即可实现高效的不对称氧化还原中性自由基环化反应. 这为未来开发新的酶催化不对称自由基反应提供了理论基础和应用方向.
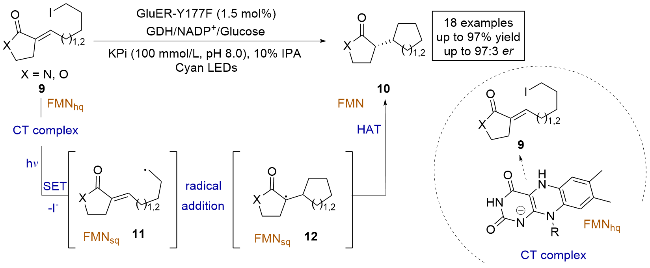
2020年, Hyster课题组[13]报道了使用活性较低的烷基碘代物作为亲核自由基前体, 进行分子内不对称自由基环加成反应(Scheme 3). 他们采用了黄素依赖的烯烃还原酶(Glu-ER)变体, 成功实现了对反应的化学选择性和立体选择性的良好控制. 紫外-可见光吸收光谱测定表明, 底物9与FMNhq在酶的存在下形成电荷转移(charge transfer, CT)复合物, 并在光照条件下激发发生单电子转移(single electron transfer, SET). 烷基碘代物通过还原脱碘生成亲核性的烷基自由基中间体11, 随后与烯烃发生分子内自由基加成, 生成前手性的碳自由基中间体12. 接着, 该中间体与还原态辅因子FMNsq发生立体选择性的HAT, 最终得到产物10. 这种策略能够高效合成手性的酯、酰胺和内酯, 扩展了可用于生物催化的还原自由基环化反应的类型.

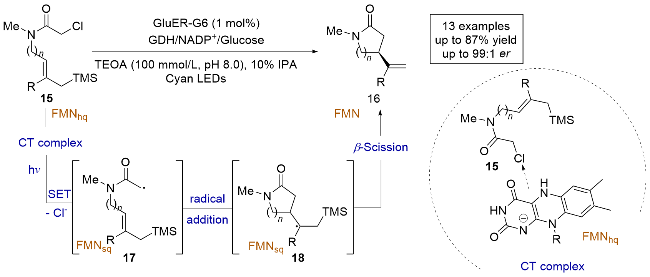
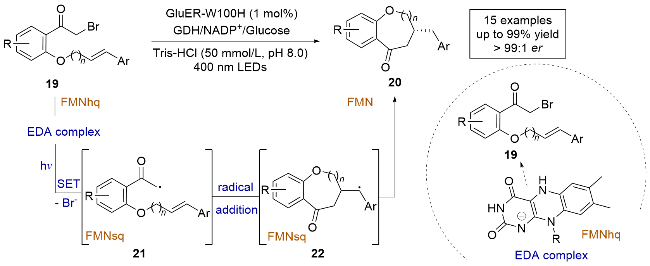
含氧的苯并稠合杂环在制药工业中是重要的结构基元, 然而, 通过自由基介导的方法进行这些化合物的不对称合成仍然是一个具有挑战性的课题. 2023年, 饶义剑课题组[16]提出了一种新颖的不对称自由基介导的光酶合成策略, 通过对黄素依赖的“烯”还原酶(GluER)进行结构引导的工程改造, 成功实现了含氧苯并稠合杂环的高效对映选择性合成(Scheme 6). 研究发现, GluER-W100H变体在可见光照射下, 能够高效地生成各种苯并氧䓬酮、色满酮和不同苯并稠合环的茚酮, 并且具有高产率和良好的立体选择性. 作者通过紫外-可见吸收光谱的测定证明了α-溴代酮类化合物19与FMNhq会生成EDA复合物, 机理研究表明, 光激发底物19和FMNhq形成的EDA复合物, 使得底物19发生还原脱溴反应, 生成自由基中间体21, 随后与烯烃进行对映选择性的自由基加成反应, 生成碳自由基中间体22, 最终通过与FMNsq的氢原子转移(HAT)反应生成目标产物. 因此, 该研究提供了一种绿色替代方法, 用于有效地化学酶合成生物活性药物的重要手性骨架.
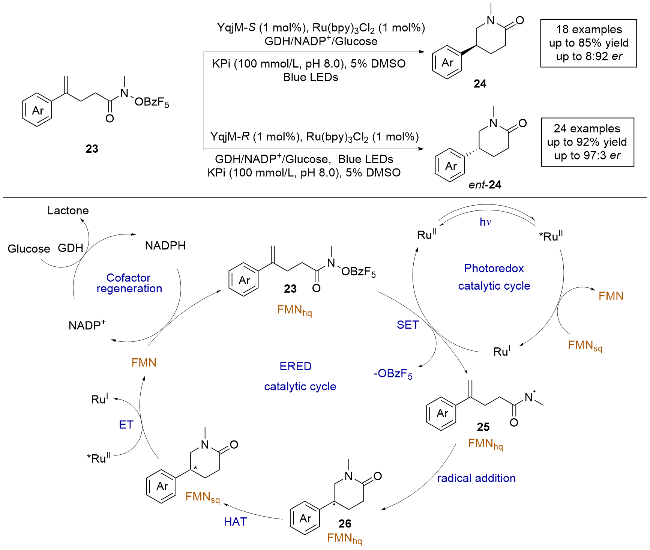
这项研究采用了酶与光敏剂协同催化的策略, 即光酶协同催化, 相较于单独的光酶催化, 该方法需要外加光敏剂辅助. 作者利用其开发的基于EREDs的光酶平台, 实现了氮中心自由基的C—N键构建. 通过定向进化, 成功控制了反应的化学选择性和对映选择性. 反应机理如下: 在蓝光照射下, 光敏剂Ru(bpy)3Cl2(RuII)被激发生成长寿命的三重态激发态配合物RuII*, 随后被FMNsq还原生成RuⅠ. 与酶结合的底物23被光敏剂RuⅠ还原, 脱去一分子的苯甲酰基生成氮中心自由基中间体25. 接下来, 自由基中间体25对分子内的烯烃进行自由基加成, 生成前手性的碳自由基中间体26. 在酶的作用下, 该中间体与FMNhq发生不对称的HAT生成目标产物. 此过程中生成的FMNsq与激发态RuII*光催化剂进行单电子转移形成FMN, FMN通过辅助因子再生系统还原为FMNhq, 从而完成光氧化还原和ERED催化循环.
1.2 单电子氧化途径
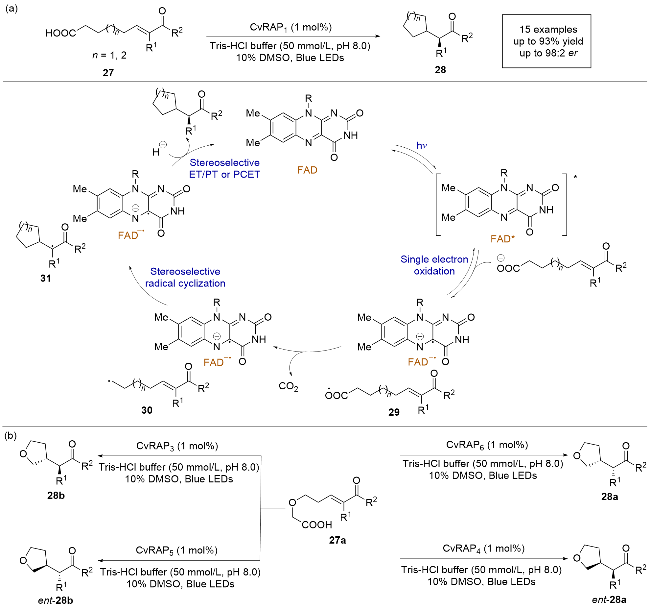
2024年, 杨扬课题组[19]报道了一例利用脂肪酸光脱羧酶(FAPs)催化的脱羧自由基环化反应. 该方法能够以高化学选择性和对映选择性构建α-手性酯(Scheme 8a), 并通过对FAPs的定向进化实现α-手性酯及具有连续两个手性中心酯的立体发散合成(Scheme 8b). 机理研究表明, FAPs的辅助因子黄素腺嘌呤二核苷酸(FAD)在光照条件下被激发, 与底物在酶的活性位点内发生单电子转移, 底物27的负离子被氧化生成氧自由基中间体29, 随后脱去一分子二氧化碳生成烷基自由基中间体30. 该中间体与烯烃发生分子内自由基加成, 生成前手性的碳自由基中间体31, 最终通过还原和不对称质子化得到目标产物. 该研究展示了利用天然光酶发展非天然反应的可能性, 拓宽了天然光酶在化学合成方面的应用范围, 为未来的研究提供了新的方向和可能性.
2 分子间自由基加成
2.1 单电子还原途径
相对于分子内反应, 分子间反应具有更容易的底物制备和更广泛的反应多样性等优势. 然而, 除了不对称光酶催化的固有困难(如不对称诱导和兼容性问题)之外, 还有几个挑战需要解决. 首先, 初始生成的亲电自由基需要具有较长的寿命, 并且能够选择性地进行交叉偶联. 其次, 这种双分子转化要求羰基和烯烃都能接近酶的活性位点, 并且位于与黄素辅因子结合位点空间上相邻的位置. 第三, 立体化学诱导更加困难, 因为新形成的C(sp3)—C(sp3)键可以自由旋转, 而中间的前手性自由基是灵活的. 这些挑战可能解释了为什么不对称光酶双分子交叉偶联反应仍然难以实现.
2020年, 赵惠民课题组[20]报道了第一例光酶催化的分子间自由基加成反应. 他们使用α-卤代酮32作为自由基前体, 烯烃33作为自由基受体, 以良好的产率和对映选择性合成γ-手性碳基化合物, 且反应表现出良好的官能团耐受性. 为了进一步了解反应机制, 作者通过测定紫外-可见吸收光谱, 表明底物和FMNhq在酶存在下形成EDA复合物, 并通过自由基钟实验证明了反应是通过自由基途径发生的. 基于所有实验数据, 作者提出了这种光酶催化烯烃氢烷基化反应的合理机制(Scheme 9): 底物与氨基酸残基通过氢键相互作用结合在酶的活性位点上, 从而与相邻的FMNhq形成EDA复合物, 之后在光照条件下激发发生分子间的单电子转移, 底物32还原脱卤生成碳中心自由基中间体35, 之后与烯烃33发生分子间的自由基加成, 得到前手性的碳自由基中间体36, 再与FMNsq发生不对称的HAT, 生成目标产物. 该反应的局限在于不能很好地控制具有两个手性中心化合物的非对映选择性. 总之, 该工作实现了两分子底物的不对称交叉偶联, 验证了光酶催化策略的可行性, 并为分子间反应提供了一种新的方法.
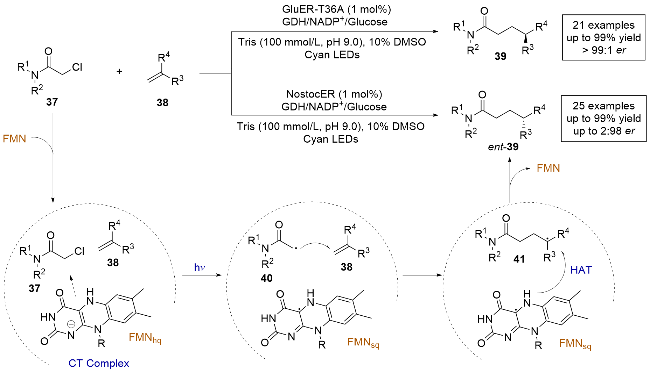
同年, Hyster课题组[21]报道了一种新颖的四元电荷转移复合物在光酶促烯烃的分子间氢烷基化反应中的作用(Scheme 10). 该方法利用黄素依赖的烯烃还原酶(Glu-ER)进行催化, 在光诱导下以极高的产率和对映选择性生成烯烃的氢烷基化产物39, 并且很少观察到传统方法中α-氯代酰胺容易脱卤氢化的副产物. 该反应表现出良好的底物官能团耐受性, 但其局限在于无法控制环外烯烃底物的对映选择性. 此外, 通过不同家族的烯还原酶(NostocER), 可以调控反应产物的对映选择性, 得到对映异构体ent-39. 与之前的报道[9]不同, 作者通过测定紫外-可见吸收光谱和理论计算, 意外发现底物37、底物38和FMNhq在酶的存在下生成了四元电荷转移复合物. 这表明自由基的生成很大程度上依赖于两个底物同时结合在酶的活性位点上, 很好地解释了为什么很少观察到底物37脱卤氢化的副产物, 确保了反应的高度选择性. 机理研究表明, 四元复合物在光照激发下, 复合物中的FMNhq与氯酰胺之间发生电子转移, 导致 C—Cl键的断裂, 生成自由基40. 然后在酶的活性位点内, 自由基与烯烃38发生自由基加成, 生成的前手性自由基中间体41再与FMNsq发生不对称的HAT, 最终生成目标产物. 这项工作通过引入光酶促的自由基生成机制和新型四元电荷转移复合物, 为生物催化领域提供了一种新的思路, 展现了潜在的科学价值和应用前景.
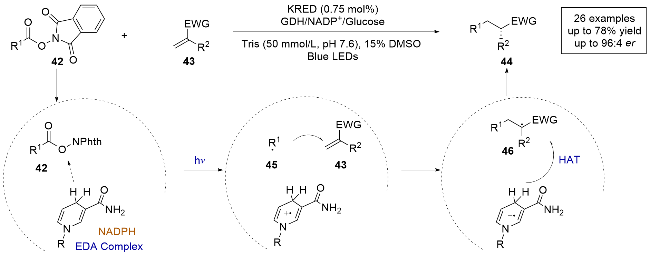
2022年, 赵惠民团队[22]报道了一例光诱导的烟酰胺依赖的酮还原酶催化的分子间自由基加成反应(Scheme 11). 不同于之前常用卤代物为自由基前体, 本研究采用易制备的氧化还原酯为底物, 与α,α-二取代末端烯烃加成生成具有α手性中心的酮类化合物, 这在传统不对称催化中是难以实现的. 酮还原酶在自然界中广泛存在, 通常用于催化生物体内的双电子反应, 这是首次将酮还原酶应用于单电子过程的双分子偶联反应. 通过对酮还原酶的半理性改造, 研究团队以优异的产率和对映选择性得到了目标产物. 通过紫外-可见吸收光谱和密度泛函理论(DFT)计算, 确认了EDA复合物的形成和光激发行为. 作者提出了这种光酶催化烯烃氢烷基化反应的合理机制: 首先氧化还原酯和烟酰胺腺嘌呤二核苷酸磷酸(NADPH)在酶的空腔内形成EDA复合物, 随后在光照激发下发生分子间的单电子转移, 氧化还原酯42被还原发生C—O键的断裂, 生成碳自由基中间体45. 接着, 自由基中间体与烯烃发生共轭加成生成前手性自由基中间体46, 随后与NADPH发生不对称的HAT生成产物. 此外, NADPH通过辅助因子循环系统缓慢再生, 确保了反应体系中NADPH的浓度保持在一个较低水平, 有效地抑制了游离NADPH引起的外消旋背景反应, 因为游离NADPH的过量会导致非选择性的自由基生成.
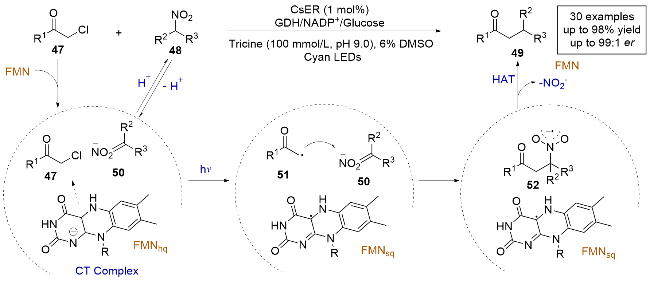
2022年, Hyster课题组[23]报道了一例α-氯代酰胺和硝基烷烃的C(sp3)—C(sp3)对映选择性亲电交叉偶联反应(Scheme 12), 该研究解决了传统金属催化剂在区分两种C(sp3)亲电试剂时的挑战, 即产率低且对映选择性难以控制的问题. 机理研究表明, 烷基卤代物47和原位生成的亚硝酸盐50在酶的存在下与FMNhq生成四元电荷转移(CT)复合物, 在光照下复合物发生电子转移, 烷基卤代物47脱卤生成自由基中间体51. 随后, 自由基中间体51与亚硝酸盐50在酶的活性位点内发生自由基加成, 生成硝基阴离子52. 之后, 在酶的介导下硝基阴离子52发生C—N键裂解, 产生亚硝酸盐和烷基自由基. 最后, 通过不对称的HAT终止反应得到对映选择性的交叉偶联产物.
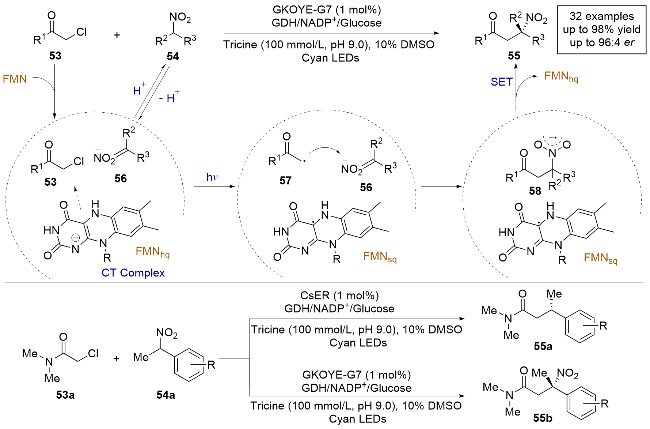
2023年, Hyster课题组[24]报道了一例利用α-氯代酰胺和硝基烷烃高化学选择性和对映选择性地实现了对硝基烷烃的C—H烷基化反应, 成功合成了高价值的手性三级硝基化合物(Scheme 13). 作者通过紫外-可见光光谱实验证实了FMNhq、α-氯代酰胺53和硝酮化合物56在酶的活性位点内形成了四元电荷转移(CT)复合物. 在光照激发下, 该四元复合物实现了FMNhq对53的单电子还原, 生成α-酰胺自由基57. 随后, 自由基对硝酮56进行立体选择性加成, 生成硝基阴离子自由基58. 最终, 自由基58被FMNsq单电子氧化得到目标产物55. 研究发现, 来自Geobacillus kaustophilus菌株的烯烃还原酶变体GkOYE-G7能够高效催化生成硝基化合物的C—H烷基化产物, 而来自Caulobacter segnis菌株的烯烃还原酶CsER则更适合催化生成亲电交叉偶联产物, 表明通过使用不同的酶可以在相同的底物条件下实现高化学选择性, 得到不同的产物, 充分体现了生物催化在化学选择性方面的优越性.
氮杂芳烃是一类含氮的芳香杂环化合物, 常作为手性生物活性分子的官能团, 广泛应用于药物、农用化学品和天然产物中. 尽管氮杂芳烃具有重要的应用价值, 但由于其平面刚性结构, 氮原子无法像羰基那样灵活地进行远程立体控制, 因此现有的方法难以区分远离单个氮杂芳烃基团的前手性面.
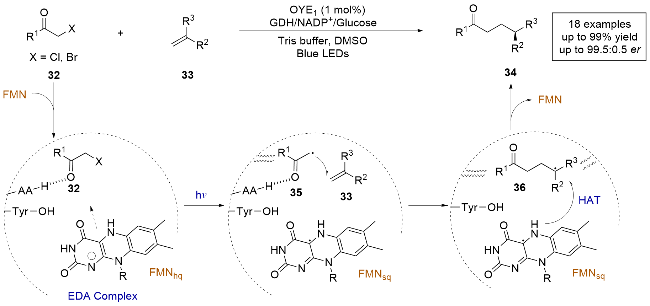
2023年, 赵惠民课题组[25]报道了一例光酶催化的溴/氯甲基取代氮杂芳烃和α-甲基取代乙烯基芳烃的分子间自由基加成反应, 实现了杂环化合物的远程立体控制(Scheme 14a). 通过采用老黄酶(OYE1), 结合对反应条件的筛选和酶的定向进化, 可以以良好的收率和对映选择性得到目标产物. 作者通过氘标记实验探索了手性HAT的来源, 并通过控制实验和理论计算提出了溴/氯甲基取代氮杂芳烃与烯烃发生氢烷基化的合理机理. 反应机理如下: 底物59和FMNhq在酶的作用下通过π-π堆积形成CT复合物, 在光照条件下激发, 底物59被还原脱卤生成氮杂苄基自由基中间体62, 随后与底物发生分子间自由基加成, 生成前手性自由基中间体63, 再与FMNsq发生不对称HAT, 最终得到产物61. 光酶催化已经显示出通过对映选择性HAT控制前手性自由基中心对映选择性的光明前景. 然而, 现有的光酶反应中通常依赖底物的羰基作为定位基团, 限制了它们在不对称催化中的应用. 该研究采用吡啶作为定位基团, 拓宽了光酶催化领域的底物范围, 为以后开发更多的光酶催化反应类型提供了新的思路.
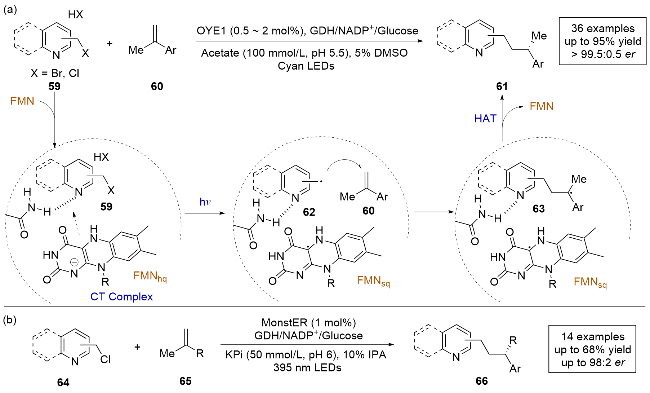
几乎同时, Hyster课题组报[26]道了一种使用黄素依赖的“烯”还原酶进行缺电子氮杂芳烃与烯烃还原偶联的光酶促反应, 也在氮杂芳烃γ-位构建了具有高立体选择性的立体中心(Scheme 14b), 不同之处在于, 作者采用了一组来自Gluconobacter属其他种类的GluER同源酶, 其中, 一种来自Gluconobacter morbifer的野生型氧化还原酶, 被命名为MonstER, 对反应有较好的催化能力, 能够以高产率和高对映选择性控制实现对杂环化合物66的远程立体控制.
2023年, 徐鉴课题组[27]报道了一例光酶催化重氮化合物和烯烃发生的不对称氢烷基化反应(Scheme 15). 在此之前, 重氮类化合物在光酶催化体系中尚未被开发. 作者利用重氮分子在水相中具有较强的氧化能力和稳定性, 可以与酶中原位生成的FMNhq发生单电子氧化还原, 再与烯烃反应. 通过对老黄酶(OYE1)的定向进化, 可以高产率和立体选择性地合成γ-手性碳基化合物, 并且能够抑制重氮化合物在光照下生成卡宾中间体与水插入或与烯烃反应的副产物. 作者通过测定紫外-可见吸收光谱证明了底物67与68和FMNhq在酶的存在下生成四元EDA复合物, 并结合自由基捕获实验证明这是一个自由基历程的反应. 根据所有的实验数据和以往的文献报道, 作者给出了合适的反应机理: 光直接激发底物在酶的作用下生成的EDA复合物, 重氮化合物被还原并伴随着氮气的离去生成烷基自由基中间体70, 之后与烯烃发生分子间自由基加成, 再与FMNsq发生不对称HAT得到产物. 作者开发了一种新的光酶催化体系中生成烷基自由基前体的底物, 与传统的卡宾介导反应相比, 这种转化提供了重氮化合物在光酶体系中的新应用, 进一步扩大了可用于光酶催化的底物类型.
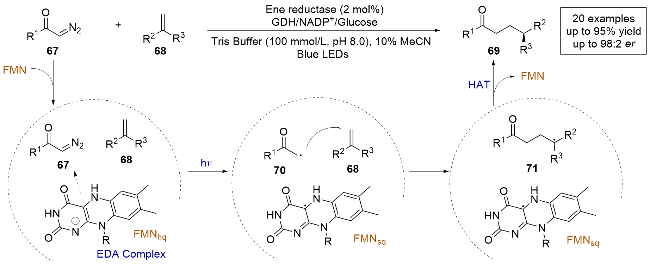
同年, 徐鉴课题组[28]又报道了一例光酶催化自由基介导的烯烃不对称氢磺酰化反应(Scheme 16). 通过对老黄酶(OYE1)定向进化, 可以合成β-手性磺酰化合物, 并且能够很好地控制反应的化学选择性和对映选择性. 他们进行了自由基捕获实验和紫外-可见吸收光谱的测定, 发现只有磺酰氯化合物和FMNhq在酶的作用下生成了EDA复合物, 烯烃并不参与. 与之前该组报道的反应机制类似[27], 光直接激发在酶作用下生成的EDA复合物, 通过电子转移过程使底物72还原脱氯, 生成砜自由基中间体75, 随后参与分子间自由基加成, 最后得到的前手性自由基中间体76与FMNsq发生不对称HAT, 生成β-手性磺酰化合物. 这项工作为在药物中普遍存在的手性磺酰基团提供了一种强有力的新方法, 并进一步扩展了黄素依赖性烯烃还原酶在光酶催化体系中的应用.
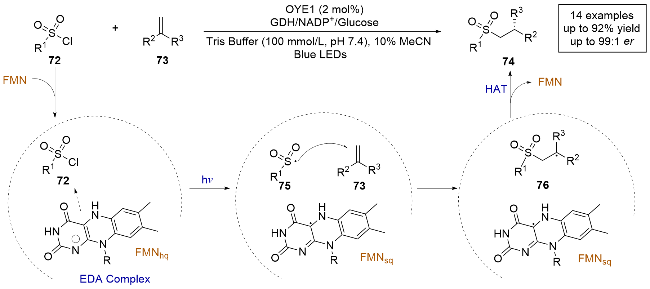
2023年, Hyster课题组[29]报道了一种由黄素依赖的烯烃还原酶催化的烯烃不对称碳羟基化反应(Scheme 17), 首次实现了通过碳羟基化反应从简单烯烃制备手性叔醇. 作者基于之前对黄素蛋白的研究, 研究了无氧条件下α-溴代苯乙酮与α-甲基苯乙烯的反应. 筛选结果显示, 来自恶臭假单胞菌的吗啡酮还原酶(MorB)能有效生成碳羟基化产物, 且只有极少的还原偶联副产物. 经过数轮迭代饱和诱变之后, 最终得到的突变体(MorB- B3)能够以高产率和高对映选择性生成目标产物, 且未检测到还原偶联副产物. 通过测定紫外-可见光吸收光谱, 发现底物并未与FMNhq生成CT复合物. 控制实验表明, MorB-B3在无光照条件下也能以较低的收率生成碳羟基化产物, 并且产物的对映选择性没有变化, 这一观察表明, FMNhq是引发反应的关键, 而光激发提供了更强的驱动力来启动自由基反应. 结合所有实验数据, 作者给出了合理的机理: 首先, α-溴代苯乙酮77与原位生成的FMNhq发生电子转移生成自由基80, 之后自由基80进攻烯烃形成中间体81. 接着, 在酶空腔中发生环化形成α-氧自由基82, 该自由基被FMNsq氧化后水解, 最终得到产物.
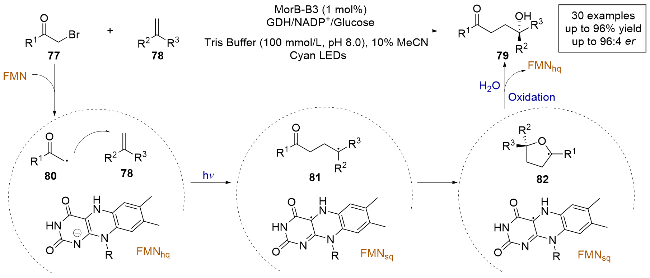
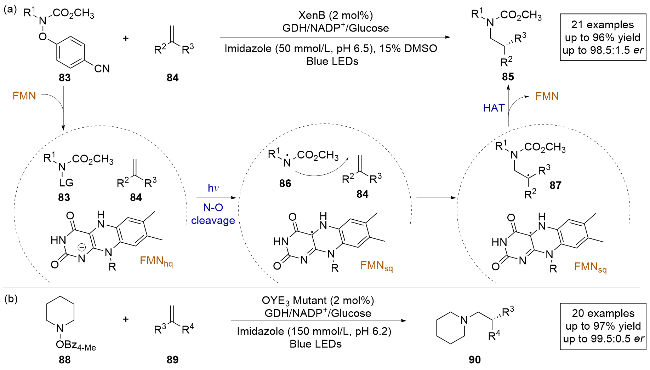
2023年, 赵惠民课题组[30]报道了一例光酶催化苯酚保护的三级胺和烯烃发生的对映选择性分子间自由基氢胺化反应(Scheme 18a). 该反应采用黄素依赖性烯烃还原酶(XenB), 因为激发态的FMNhq是一种强大的还原剂, 可以直接与底物发生电子转移, 而无需额外添加光催化剂或还原剂. 采用苯酚作为保护基团的含氮底物, 因为N—O键不稳定, 相对容易被还原发生均裂生成氮自由基中间体, 且这些含氮底物的还原电位可以通过改变离去基团上的取代基来调节. 作者通过对照实验发现, pH值和缓冲液的类型是影响该反应结果的关键因素, 且反应在酸性条件下通常得到更好的产率, 这可能是因为在酸性条件下更容易中和离去的苯酚负离子. 通过对来自恶臭假单胞菌烯还原酶(XenB)的定向改造, 可以以极高的收率和对映选择性得到产物. 作者还进行了自由基钟实验, 证明该反应是通过自由基途径发生的. 通过稳态和时间分辨光谱测量, 观察到酶的吸收特征并不随底物浓度而变化, 表明没有形成EDA复合物. 结合氘代实验和理论计算, 证明了反应的终止步骤是黄素和底物的HAT步骤. 基于所有实验数据, 作者提出了合理的反应机理: 含氮底物83在酶的活性位点在光照条件下被FMNhq还原, 脱去苯酚负离子生成氮自由基中间体86, 随后与烯烃发生分子间的自由基加成, 生成前手性自由基中间体87, 再通过不对称HAT得到氢胺化产物. 该工作清楚地展示了生物催化和光催化相结合的潜力, 可以解决合成化学中的长期挑战, 并为新型合成方法的发展带来更多的多样性. 之后, 该课题组[31]又报道了类似的工作, 采用氮的α-位带有氢原子的底物88和烯烃89进行氢胺化反应(Scheme 18b), 用老黄酶催化, 避免发生氮中心自由基的1,2-氢迁移生成副产物, 最终以高化学选择性和高对映选择性得到氢胺化产物.
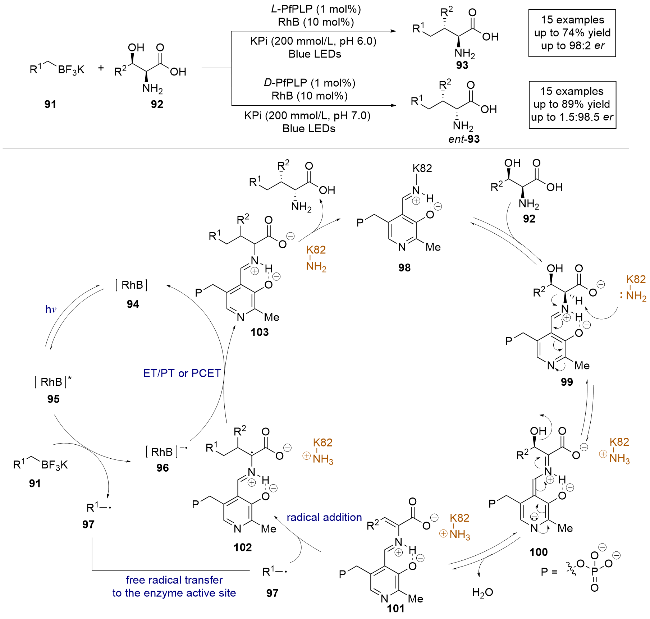
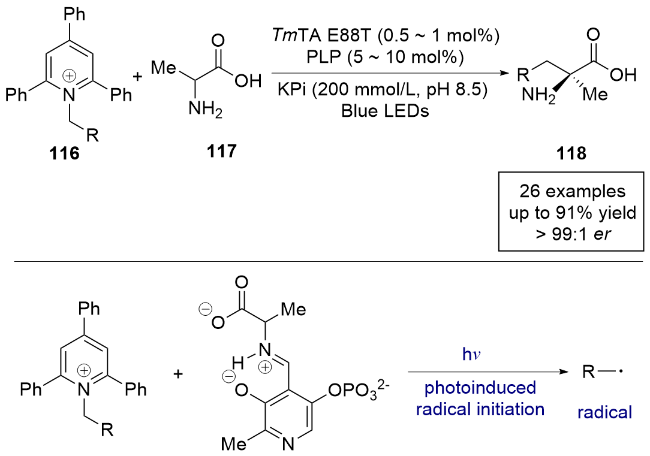
2023年, 杨扬课题组[32]报道了一例光酶协同催化烷基三氟硼酸钾盐和天然氨基酸的分子间自由基加成反应, 实现了天然氨基酸的β-烷基化(Scheme 19). 该研究使用5'-磷酸吡哆醛(PLP)依赖性酶, 通过对酶进行定向进化和反应条件的优化, 以高产率实现了具有单立体手性中心的非天然氨基酸的L/D型调控, 甚至构建了具有两个或三个相邻手性中心的非天然氨基酸. 控制实验表明, 该反应仅在有光、光敏剂和酶的共同作用下发生. 通过一系列控制实验和理论计算, 作者给出了合理的反应机理: 首先, 可见光照射光氧化还原催化剂94产生激发态光催化剂95, 烷基三氟硼酸盐底物91在激发态光催化剂作用下进行单电子氧化, 生成碳中心自由基97和还原态光催化剂96. 同时, PLP依赖性β-功能化酶98 (色氨酸合成酶)与氨基酸底物92发生转氨反应, 形成外部醛亚胺99, 从而显著降低羧基α位质子的pKa, 易于去质子化形成关键的醌类中间体100, 继而脱水生成烯烃中间体101. 在此阶段, 光催化形成的自由基97进入酶的活性位点, 进行对映选择性的分子间自由基加成, 生成以碳为中心的自由基102, 随后与还原态光催化剂发生电子转移(ET)、质子转移(PT)或质子耦合电子转移(PCET), 最终释放产物并再生PLP依赖性酶, 完成整个催化循环. 该光氧化-吡哆醛自由基生物催化协同体系为控制自由基的不对称催化反应提供了新的思路.
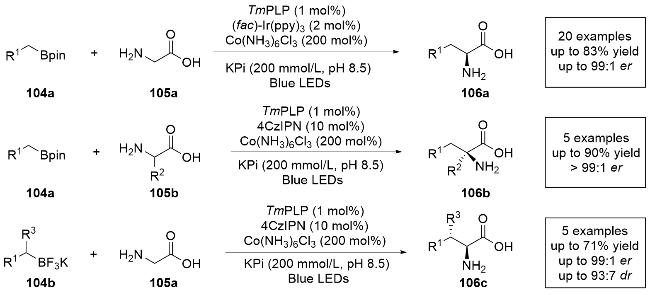
2024年, 杨扬课题组[33]再次报道了一例通过光酶催化有机硼试剂和氨基酸的分子间氧化偶联, 实现天然氨基酸的α-烷基化反应(Scheme 20). 利用5'-磷酸吡哆醛(PLP)依赖性的苏氨酸醛缩酶, 通过自由基机制对甘氨酸和α-支链氨基酸底物进行α-碳氢官能团化, 产生了一系列α-三取代和四取代的非天然手性氨基酸. 需要注意的是, 生成α-三取代和四取代的非天然手性氨基酸所用的光敏剂是不同的. 此外, 该方法还能够以高非对映选择性和对映选择性合成具有两个连续立体中心的产物. 通过对苏氨酸醛缩酶的定向进化, 该酶能够耐受初级和次级自由基前体, 包括苄基、烯丙基和烷基硼试剂. 控制实验表明, 氧化剂的加入可以显著提升反应产率, 这是因为氧化剂可以循环反应体系中的外加光敏剂. 他们成功地进行了对映选择性保持的毫摩级别106的制备, 证明了该方法的合成效用. 反应机理与之前报道[32]类似, 不同之处在于生成的氮自由基中间体被光催化剂氧化去质子, 生成氧化偶联产物. 该研究为氧化交叉偶联提供了新的方法, 拓宽了光酶催化体系中自由基反应路径的类型.
2024年, Hyster课题组[34]报道了一例光酶催化Katritzky吡啶鎓盐与氨基酸的分子间自由基加成反应(Scheme 21). 该研究采用5'-磷酸吡哆醛(PLP)依赖的苏氨酸醛缩酶和玫瑰红光氧化还原催化剂, 通过吡啶鎓盐对未保护的氨基酸进行α-烷基化, 高产率地制备了各种手性α-取代氨基酸. 通过紫外-可见光谱阐明了吡啶鎓盐、苏氨酸醛缩酶和光催化剂之间的三元相互作用. 机理研究表明, 在光照激发酶存在下, 吡啶鎓盐107被还原生成碳自由基中间体110, 同时PLP酶活化氨基酸生成亚胺中间体112, 伴随质子离去生成醌类中间体113. 自由基110与113进行对映选择性自由基加成, 之后与光敏剂发生电子转移生成目标产物, 从而实现天然氨基酸向非天然氨基酸的高效、高选择性转化.
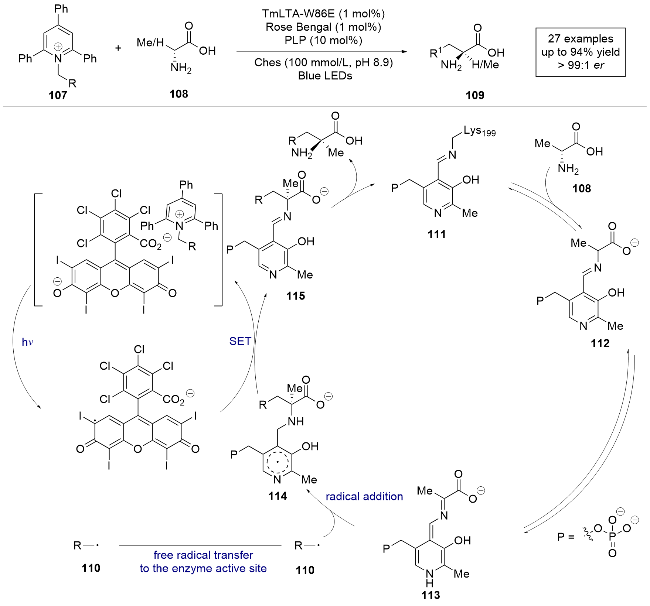
同年, 杨扬课题组[35]利用相似的策略, 以Katritzky吡啶鎓盐作为自由基前体, 通过PLP依赖性苏氨酸醛缩酶催化氨基酸的不对称α-烷基化反应(Scheme 22). 通过对苏氨酸醛缩酶的定向进化, 可以从无保护的氨基酸底物合成具有挑战性的α-三取代和α-四取代的非天然氨基酸产物, 并且具有极高的收率和对映选择性. 该研究与之前的工作不同[32-34], 无需外加光敏剂和氧化剂. 作者通过EPR实验和自由基捕获实验, 证明了Katritzky吡啶鎓盐和PLP衍生醛亚胺在光诱导下生成自由基. 这种无需经典光催化剂或光致酶的催化反应, 为在有机化学或生物化学中实现立体选择性的自由基反应开辟了新的方向.
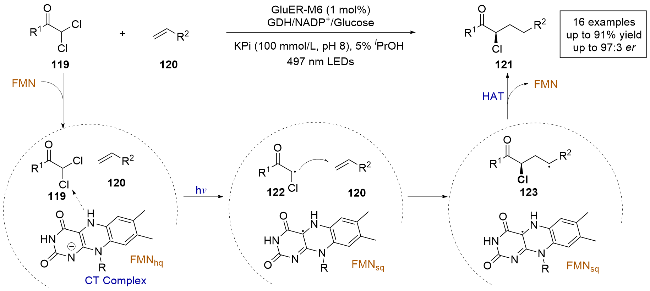
2024年, Hyster课题组[36]报道了一例光酶催化的α,α-二氯酰胺与芳基烯烃的分子间自由基环加成反应(Scheme 23). 通过对黄素依赖烯还原酶(Glu-ER)的定向进化和反应条件的优化, 以优异的化学选择性和对映选择性得到了α-氯酰胺, 并展示了良好的底物耐受性. 由于α-氯酰胺作为具有药学价值的结构单元, 作者还对其进行了后续的官能团修饰. 通过一系列控制实验以及紫外-可见吸收光谱的测定, 结合该研究中的所有实验结果, 作者提出了合理的反应机理. 首先, 自由基122通过光激发两种底物和FMNhq生成的三元EDA复合物, 经单电子还原形成, 随后对烯烃120进行对映选择性自由基加成生成自由基中间体123, 然后与FMNsq发生HAT得到产物121. 酶对电荷转移复合物内分子方向的精确控制可能是能够很好控制产物对映选择性的原因.
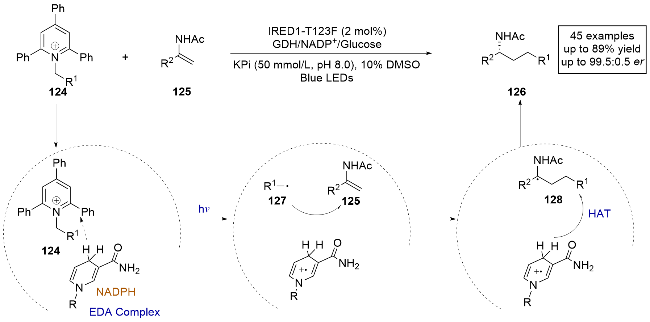
催化剂的发展是合成创新的基石, 光酶的发现就是一个很好的例子. 然而, 通过直接光激发催化新型光生物转化的天然酶种类仍然局限于黄素蛋白和酮还原酶. 2024年, 黄小强课题组[37]报道了一种亚胺还原酶(IREDs), 这种酶能够催化远程C(sp3)—C(sp3)键的形成, 提供了一种先前难以实现的烯酰胺自由基氢烷基化方法, 用于获取手性胺类化合物(45个例子, 最高可达99% ee) (Scheme 24). 这种方法具有广泛的底物适用性, 包括氧化还原酯等多种自由基前体. 除了催化两电子还原胺化反应中的天然功能外, 通过直接可见光激发或与合成光氧化还原催化剂协同作用, IREDs被重新用于调整非天然光诱导的单电子自由基过程. 为了探索催化机制, 作者进行了紫外-可见光吸收光谱和自由基捕获实验, 并基于实验和理论计算提出了合理的机理解释. 在酶的作用下, 光直接激发底物124和NADPH形成的EDA复合物, 底物124发生单电子还原生成自由基中间体127. 该中间体随后与烯烃125进行分子间自由基加成, 并通过与氧化态的NADPH发生不对称HAT反应, 最终生成产物. 该研究展示了IREDs在光酶催化中的潜力, 为有机合成中的高选择性自由基反应提供了新的方法和思路.
氟元素具有独特的电子特性、小尺寸和脂溶性, 对有机分子的功能特别是生物活性化合物有深远影响. 引入含氟基团可以增强分子的生物利用度、代谢稳定性以及与蛋白质的相互作用, 因此氟化合物在制药和农化工业中占据重要地位. 然而, 自然界中仅有极少数酶能够利用氟化底物, 限制了氟化化合物的生物合成. 传统化学方法虽然可以引入多种复杂的氟化基团, 但在远离手性中心的氟化基团上实现高对映选择性依然是一个挑战.
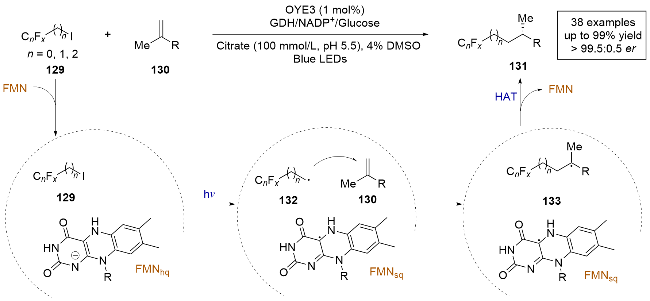
2024年, 赵惠民[38]报道了一例光酶催化的氢氟烷基化反应(Scheme 25). 该反应在光照条件下, 以三氟碘乙烷作为氟试剂, 在老黄酶(OYE3)的催化下与烯烃反应, 生成了具有高对映选择性的产物. 此方法对多种底物具有广泛适用性, 能够在多种烯烃上引入氟基团, 且收率和选择性良好. 这一方法不仅拓展了酶催化氟化反应的范围, 还成功地在远离氟取代基的β、γ或δ位置上实现高对映选择性的合成, 这是传统化学催化难以达到的. 通过控制实验和理论计算, 作者阐明了反应机理. 首先, 三氟碘乙烷在光照条件下与FMNhq发生单电子转移, 还原脱碘生成含氟碳自由基中间体132. 随后该中间体在酶的活性口袋内, 和烯烃发生分子间的自由基加成, 生成前手性自由基中间体133. 最终, 中间体133与FMN的自由基半醌中间体FMNsq发生不对称的HAT, 生成最终产物. 这一研究不仅在酶催化领域取得了突破, 也为氟化合物的合成开辟了新的途径.
2.2 单电子氧化途径
2023年, 黄小强课题组[39]报道了一例富电子芳烃与烯烃发生的分子间加氢芳基化反应(Scheme 26). 通过对酶(Glu-ER)的定向进化和反应条件筛选, 可以以优异的产率和对映选择性得到氢芳基化产物. 利用不同家族的烯烃还原酶(OYE1), 可以实现产物的立体发散合成. 该方法具有较好的底物耐受性, 对于吲哚、吡咯甚至带有给电子基团的吡啶等底物均适用. 该工作无需外加辅助因子循环系统. 通过一系列控制实验和理论计算, 作者提出了合理的反应机理: 在光照条件下, 与烯还原酶结合的FMN和富电子芳烃发生电子转移, 生成芳基自由基阳离子中间体137和半醌态黄素辅因子FMNsq, 随后, 中间体137与烯烃135发生分子间的自由基加成, 生成前手性自由基中间体138. 最后, 前手性自由基中间体与FMNsq发生HAT, 生成对映体富集的产物并再生基态FMN. 该研究通过直接可见光激发的烯还原酶实现单电子氧化引发的自由基机制, 使烯还原酶转化为光氧化剂并催化反应, 所开发的光酶活化模式有望扩展更多天然酶参与的非天然反应.
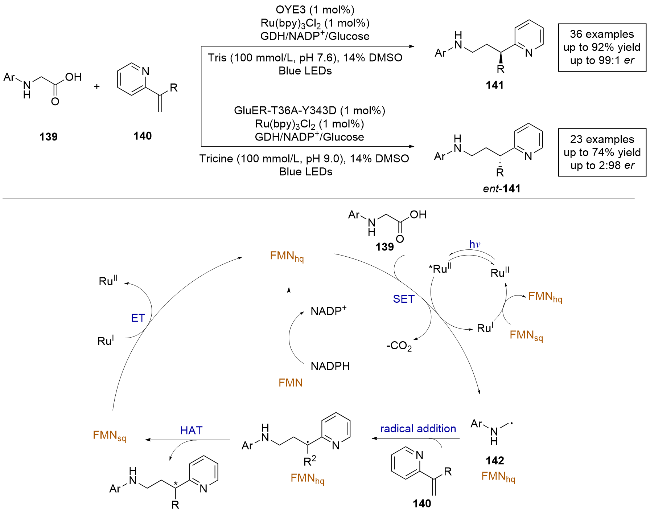
2023年, Hyster课题组[40]报导了一例利用光酶协同催化的对映体选择性脱羧烷基化反应(Scheme 27). 该反应采用黄素依赖的烯烃还原酶(OYE3)和外加光敏剂, 在光照条件下实现了氨基酸139对乙烯基吡啶140的不对称自由基加成反应, 生成产物具有优异的产率和对映选择性, 并且表现出良好的官能团耐受性. 此外, 通过定向进化不同家族的烯烃还原酶(Glu-ER), 可以实现产物的立体发散性合成. 斯特恩-沃尔默淬灭(Stern- Volmer quenching)实验表明, 在有酶和没有酶的情况下淬灭常数变化可以忽略不计, 这表明酶并没有加速羧酸的氧化. 通过理论计算和自由基捕获实验, 证明了这是一个自由基历程反应. 结合实验数据, 作者提出了对映体选择性脱羧烷基化反应的合理机理: 首先, 利用外加的GDH/Glucose/NADP+将FMN还原为FMNhq. 随后, 氨基酸底物139和乙烯基吡啶140与蛋白质活性位点结合, Ru(bpy)32+被光激发到激发态, 激发态光敏剂氧化脱羧139, 生成α-氨烷基自由基中间体142. 中间体142在酶的活性位点内与乙烯基吡啶发生分子间的自由基加成, 生成前手性碳自由基中间体. 最后, 前手性碳自由基中间体与FMNhq发生不对称HAT, 得到目标产物. 与此同时, 生成的FMNsq与光敏剂发生氧化还原反应, 再生FMNhq和光敏剂. 需要说明的是, 该工作额外加入了辅助因子循环系统, 对于酶来说, 反应的第一步是还原过程.
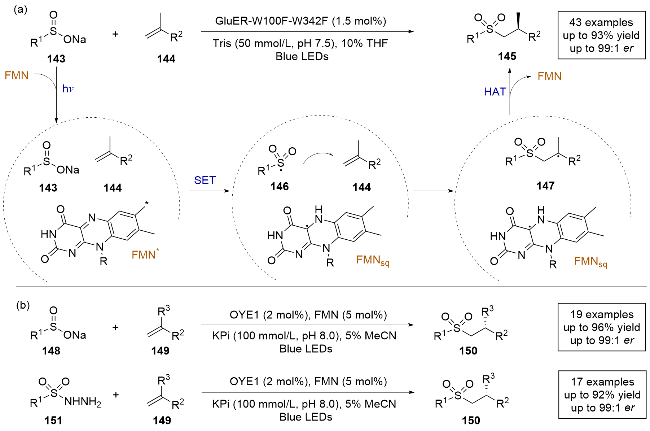
2024年, 叶俊涛课题组[41]报道了一例使用芳基亚磺酸钠和α-甲基烯烃发生的不对称氢磺酰化反应(Scheme 28a). 该研究与之前光酶催化体系中生成磺酰基自由基的方式不同[28], 通过单电子氧化生成自由基, 拓宽了自由基前体的范围, 且无需外加辅助因子循环系统, 从而简化了反应体系. 使用黄素依赖性的烯烃还原酶突变体, 可以很好地控制反应的化学选择性和对映选择性. 该方法适用的底物范围广泛, 对于芳基和烷基取代的磺酸钠均可以顺利得到产物, 并且烯烃底物的芳环可以拓展到含氧、含硫杂环. 作者还进行了自由基捕获实验, 证明该反应通过自由基路径发生. 根据实验, 作者提出了合理的反应机理: FMN在光照条件下被激发, 激发态FMN氧化芳基亚磺酸钠143生成自由基中间体146. 随后该自由基在酶的活性位点与烯烃144发生自由基加成, 生成前手性自由基148, 最终通过FMNsq发生不对称的HAT, 得到目标产物, 并再生基态FMN. 另一种可能是, 与近端酪氨酸残基发生对映选择性HAT, 生成产物后酪氨酸自由基再与FMNsq反应, 再生FMN和酪氨酸. 机理实验证明, 后者的可能性更大.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
几乎同时, 徐鉴课题组[42]也报道了一种通过FMN(黄素单核苷酸)引发的光酶催化氧化还原中性自由基氢磺酰化反应(Scheme 28b). 反应机制包括: FMN被激发后从底物获取电子, 生成磺酰自由基, 然后这些自由基与烯烃结合, 通过FMNsq (FMN半醌态)进行氢原子转移, 从而完成反应. 该方法的底物适用范围更加广泛, 不仅使用芳基或烷基取代的亚磺酸钠作为自由基前体, 还拓展至芳基或者烷基取代的磺酰肼. 此外, 与之前关于由黄素依赖酶催化的光诱导非天然反应大多依赖于还原路径的研究不同, 该方法还克服了牺牲电子供体的局限性. 不仅提高了反应的化学经济性, 还扩展了光生物催化中的反应模式.
3 总结与展望
近年来, 利用光酶催化策略实现自由基不对称加成反应取得了显著进展. 光酶催化通过绿色温和的方式, 高效合成手性化合物, 展示出在对映选择性氢原子转移(HAT)控制方面的卓越性能, 弥补了传统不对称催化方法在这一领域的不足. 该策略不仅实现了高对映选择性的产物合成, 还通过合理的酶工程和反应优化, 为复杂化学反应提供了新的途径. 然而, 目前的光酶反应主要依赖底物的羰基作为定位基团, 导致底物种类受限, 难以涵盖更广泛的化学结构. 因此, 未来的研究应致力于开发更多种类的定位基团, 例如含氮、含硫等不同类型的官能团, 以拓展光酶催化的应用范围, 增强其在复杂分子合成中的通用性和灵活性.
此外, 目前可用于光酶催化不对称自由基加成反应的酶种类相对有限, 限制了其在更广泛化学反应中的应用. 亟需开发和优化更多的天然酶和人工酶, 通过定向进化和酶工程手段, 增强其对非天然底物的催化能力. 现有的研究主要集中在构建中心手性产物, 而未来可以进一步拓展到轴手性和面手性等更复杂的手性化合物的构建, 如螺环、环状以及桥环化合物等. 这不仅能够丰富手性化合物的种类, 还能为材料科学、药物化学和农用化学品的研发提供更多的选择.
本综述致力于填补光酶催化不对称自由基加成反应的研究空白, 为该领域提供新的研究视角和理论支持. 希望通过不断的研究和创新, 进一步拓展光酶催化反应的应用范围, 推动其在绿色化学和高效合成中的进一步发展, 最终实现从实验室到工业应用的转化.
(Cheng, F.)