


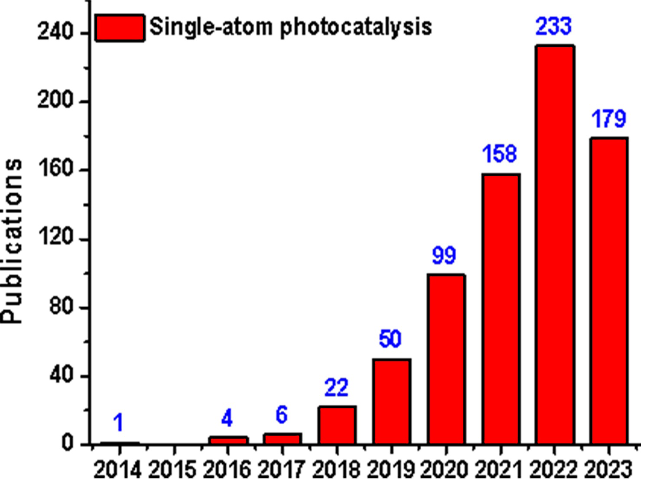
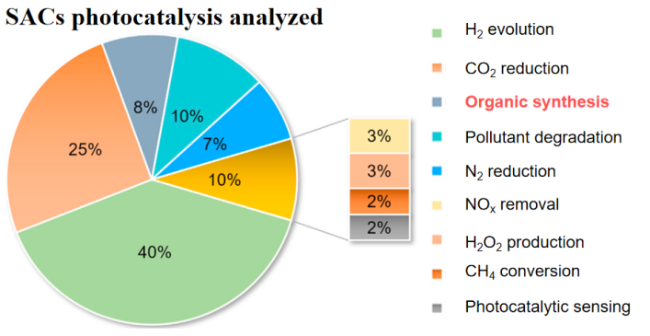
1 单原子光催化构建C—N键


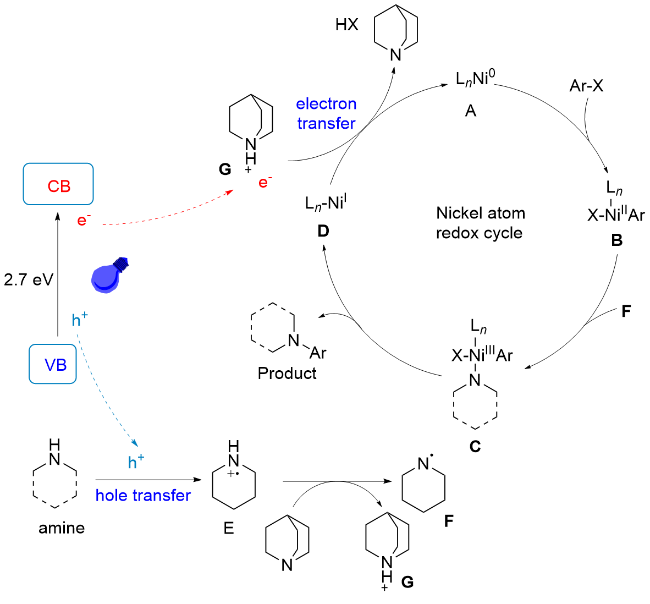

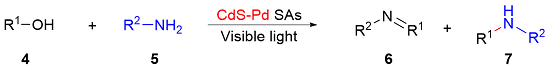
表1 光催化胺与醇的N-烷基化反应Table 1 Photocatalytic N-alkylation of amines with alcohols |
| Entry | Alohols | Amines | Product | Amine | Yield/% | |
|---|---|---|---|---|---|---|
| 6 | 7 | |||||
| 1 | 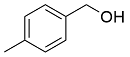 | 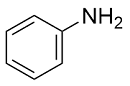 | 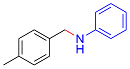 | 90 | 21 | 69 |
| 2 | 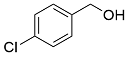 | 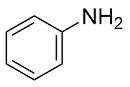 | 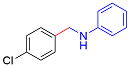 | 65 | 21 | 44 |
| 3 | 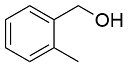 |  | 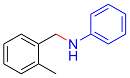 | 88 | 18 | 70 |
| 4 | 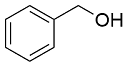 | 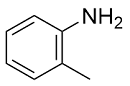 | 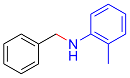 | 91 | 19 | 72 |
| 5 | 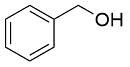 | 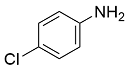 | 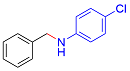 | 70 | 29 | 41 |
| 6 | 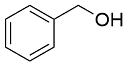 | 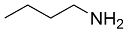 | 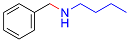 | 31 | 15 | 16 |
| 7 | 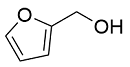 | 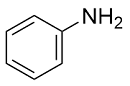 | 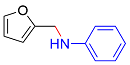 | 62 | 23 | 39 |
| 8 | 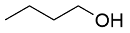 | 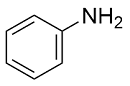 | 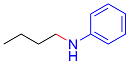 | 26 | 12 | 14 |
| 9 | 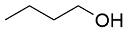 | 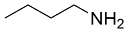 | 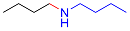 | 22 | 13 | 9 |
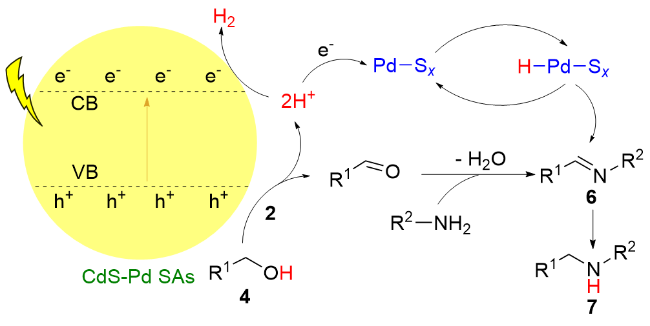

图5 (A) Ag/g-C3N4的TEM图像和(B) g-C3N4、块状g-C3N4、Ag NPs和Ag/g-C3N4复合材料的紫外光谱Figure 5 (A) TEM images of Ag/g-C3N4, and (B) UV-vis spectra of g-C3N4, bulk g-C3N4, Ag NPs, and Ag/g-C3N4 composites Copyright 2016 Wiley-VCH. |

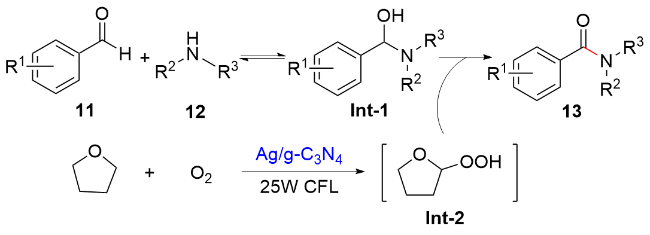
图式4 Ag/g-C3N4催化芳香醛和胺氧化酰胺化Scheme 4 Oxidative amidation of aromatic aldehydes and amines catalyzed by Ag/g-C3N4 |
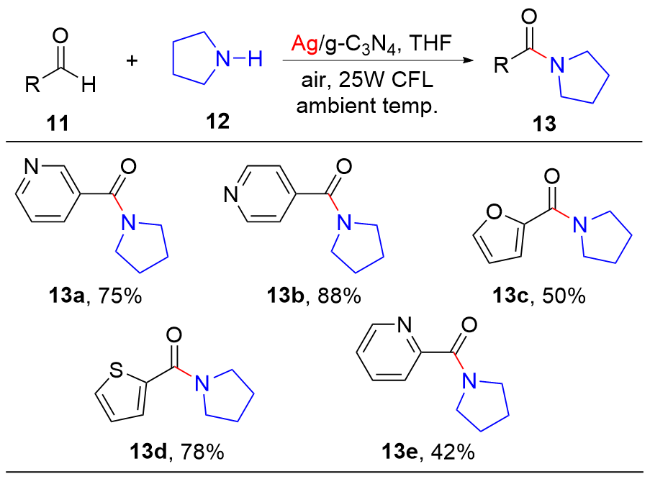
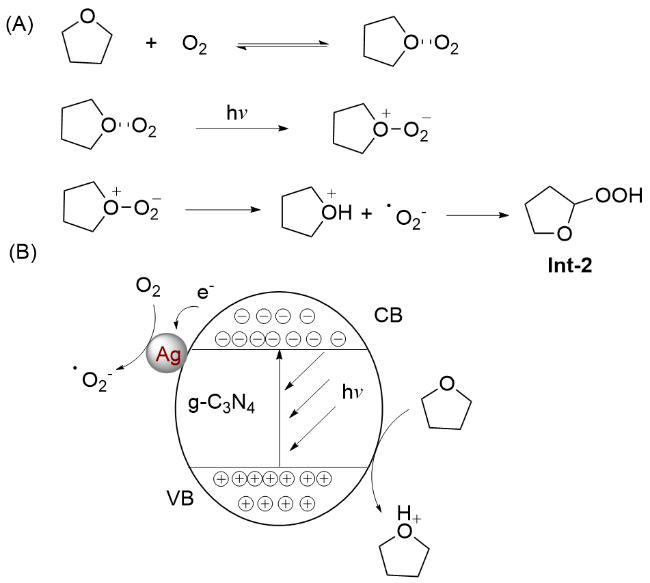
图式6 (A)光催化四氢呋喃的氧化机理和(B)可见光照射下Ag/g-C3N4表面的电子转移Scheme 6 (A) Mechanism of photocatalytic oxidation of THF, and (B) Electron-transfer approach on Ag/g-C3N4 surface under visible light irradiation |

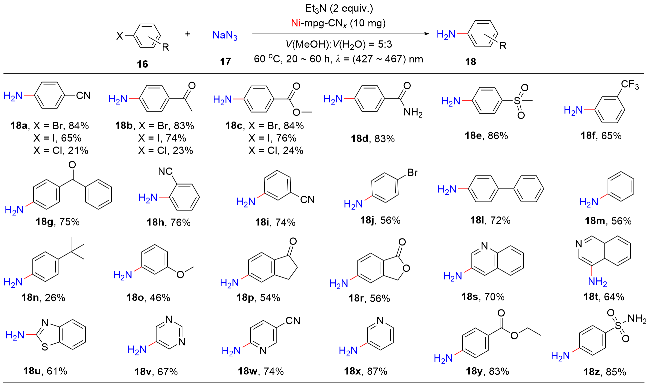
2 单原子光催化构建C—O键

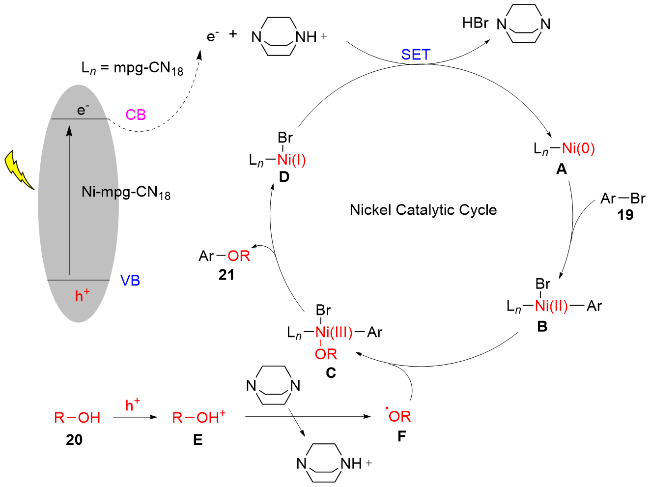
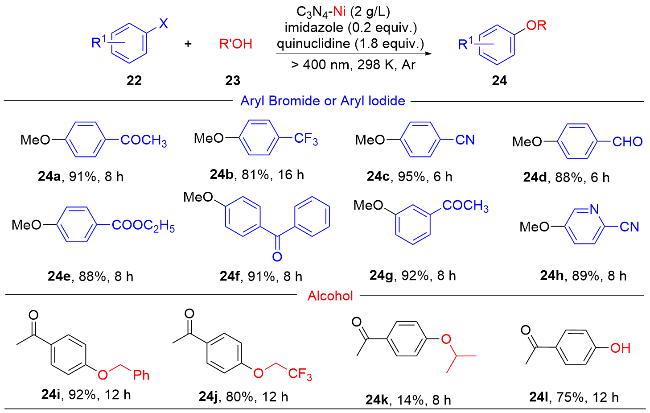
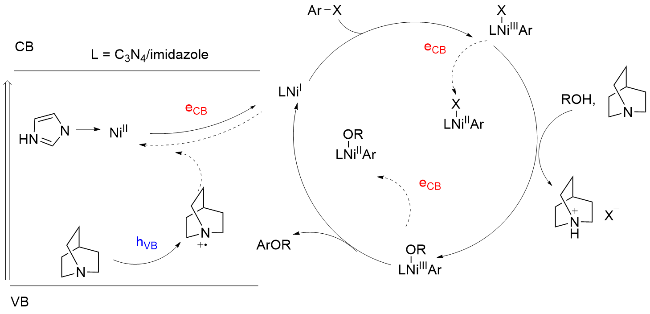
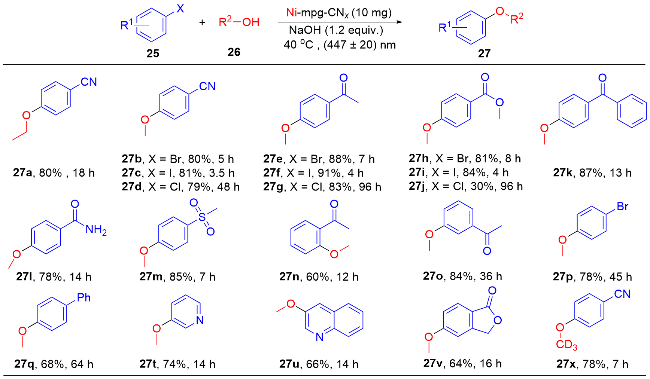
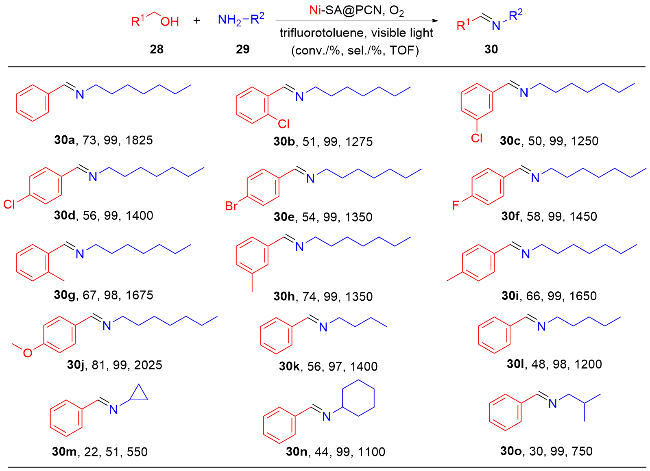
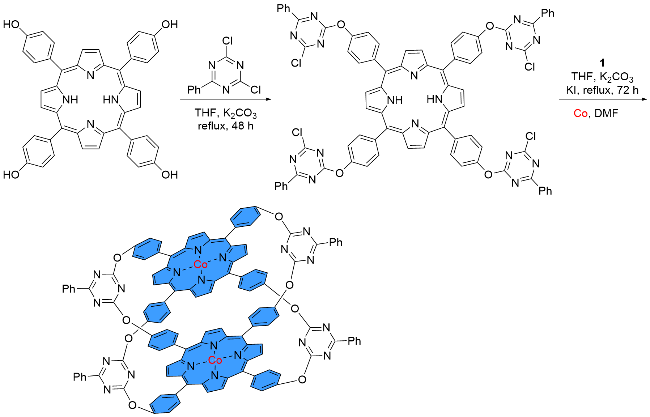
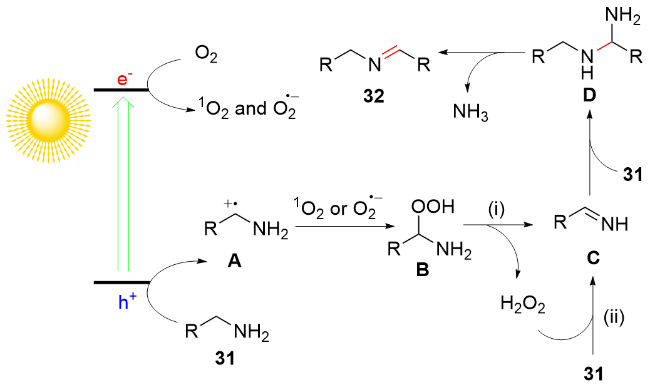
3 单原子光催化构建C=N键

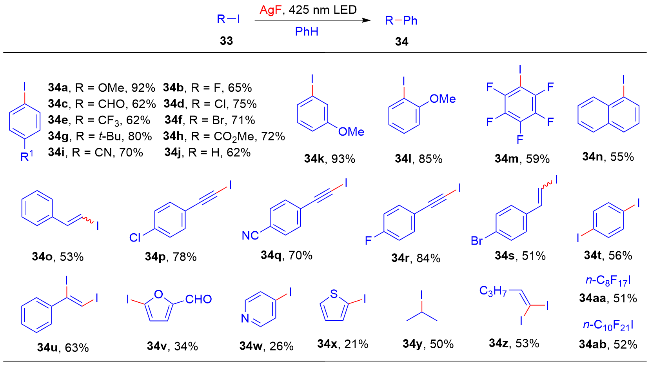
4 单原子光催化构建C—C键
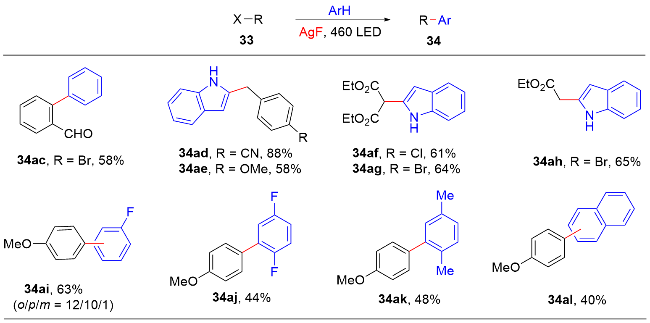
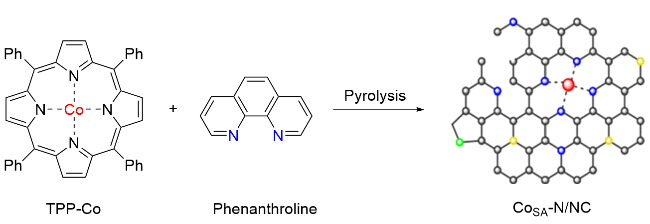
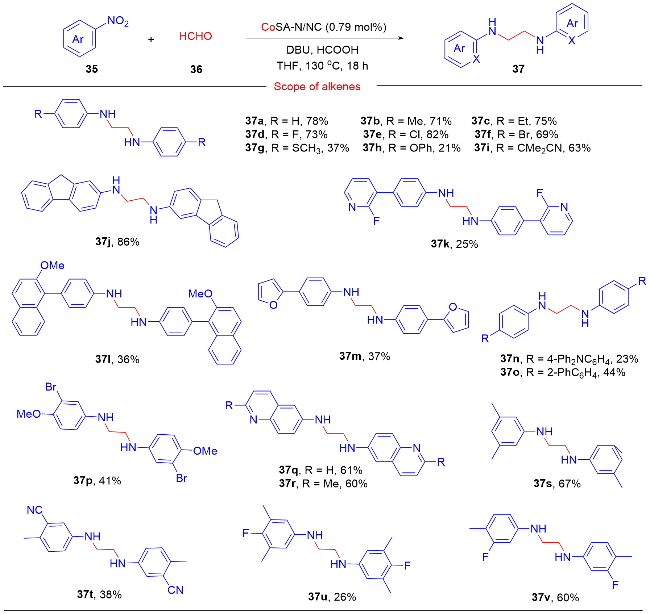
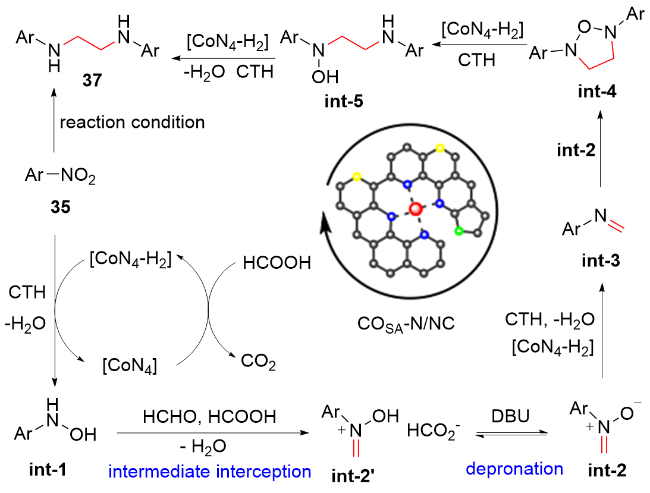
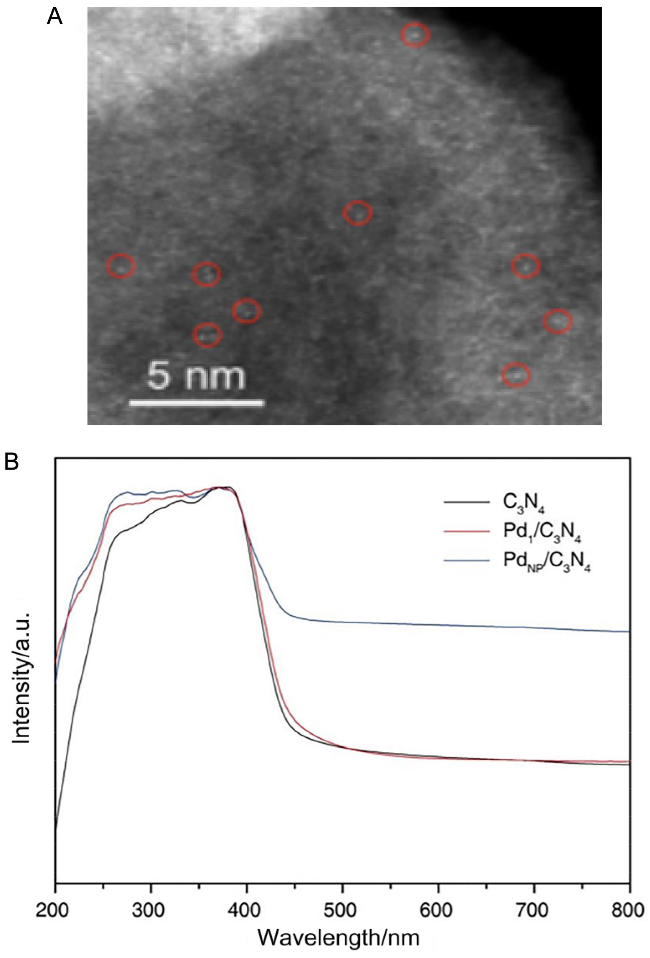
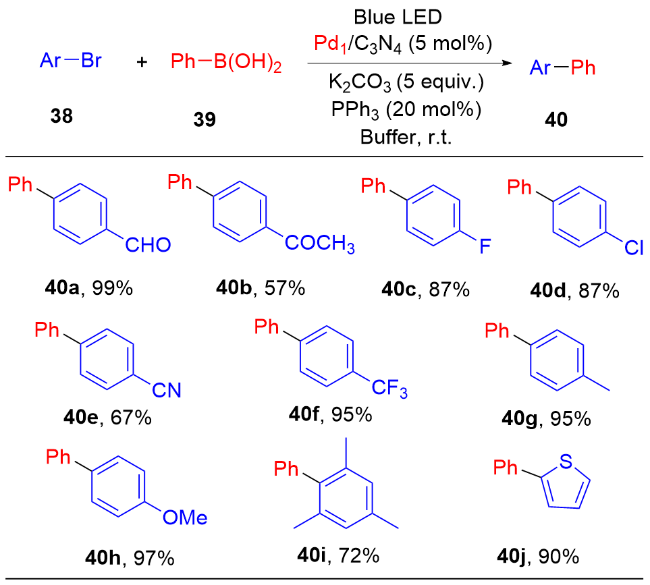
5 单原子光催化构建C—S键
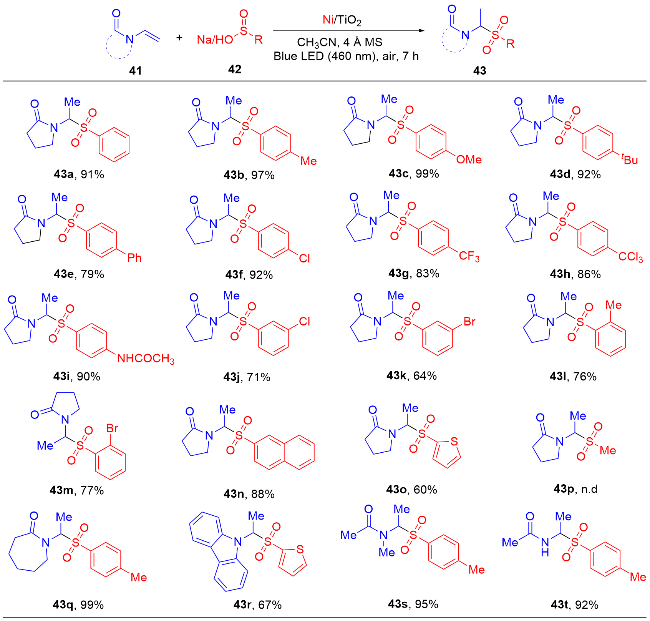
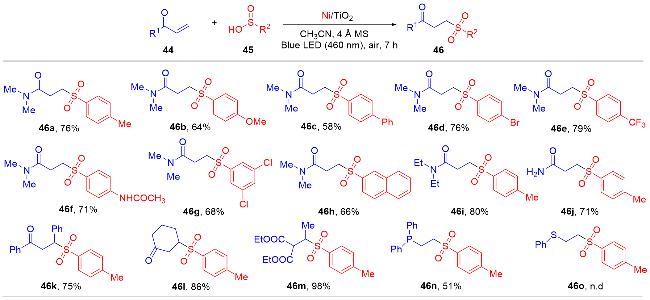
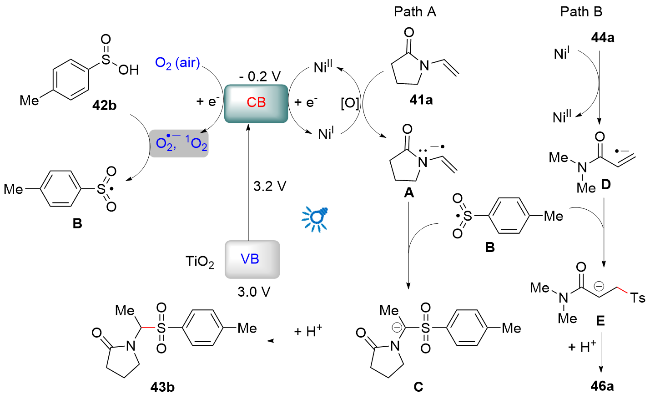

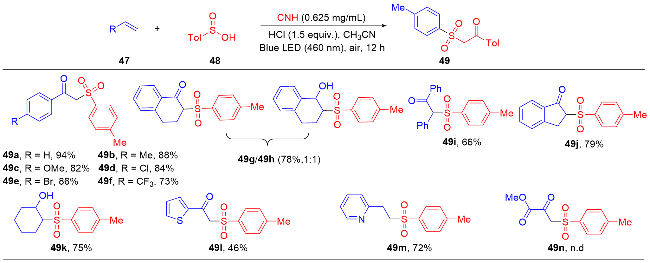
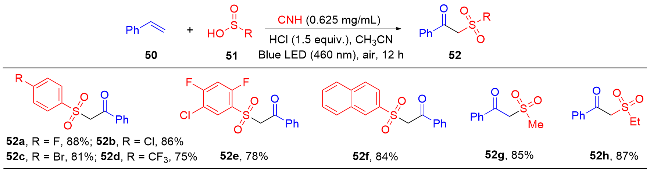
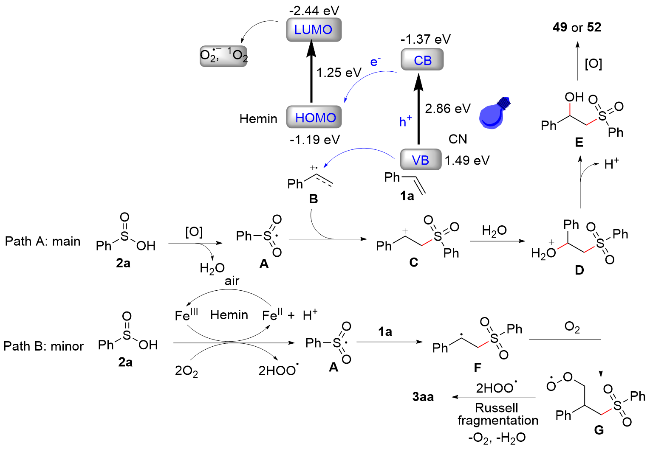
6 单原子光催化构建C—B键
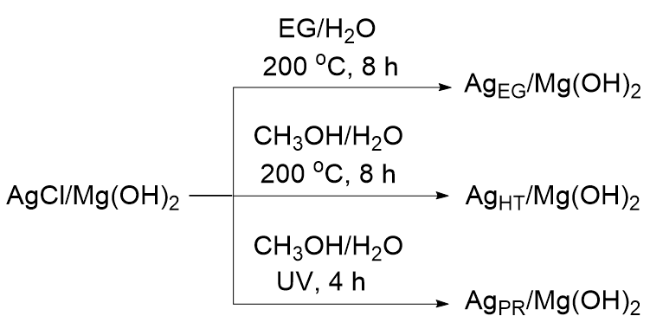

图9 (A) Ag(0)/Mg(OH)2的高角环形暗场像差校正图像和(B) Ag (0)/Mg(OH)2和Mg(OH)2的紫外吸收光谱Figure 9 (A) High-angle annular dark field aberration corrected (HAADF AC)-STEM image of Ag(0)/Mg(OH)2, and (B) UV-vis spectra of Ag(0)/Mg(OH)2 and Mg(OH)2 Copyright Applied Catalysis B: Environmental 299 (2021) 120674. |
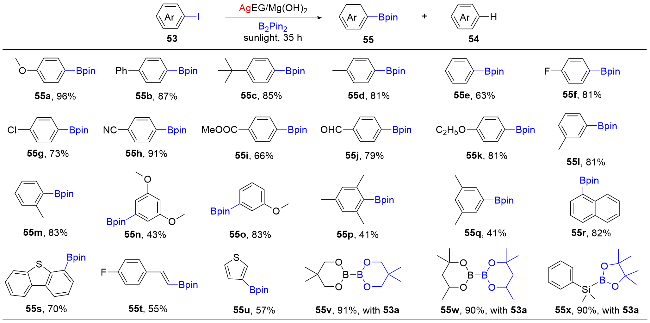
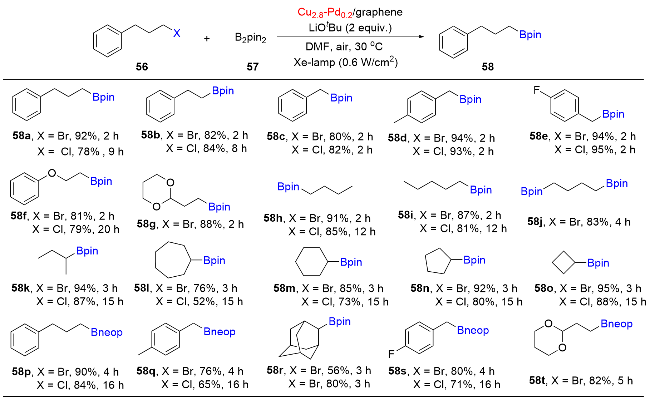
7 单原子光催化构建C—P键



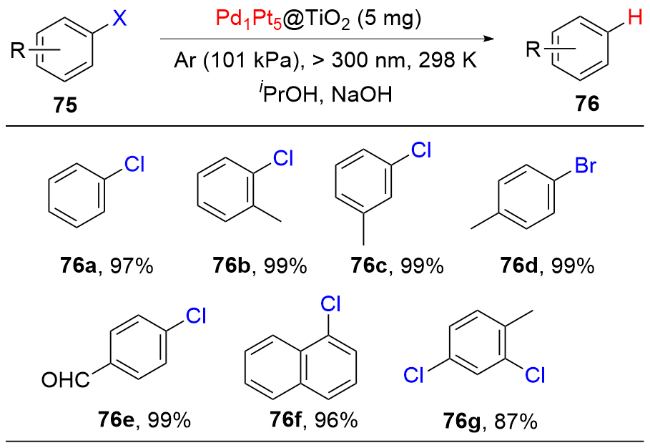
8 单原子光催化还原
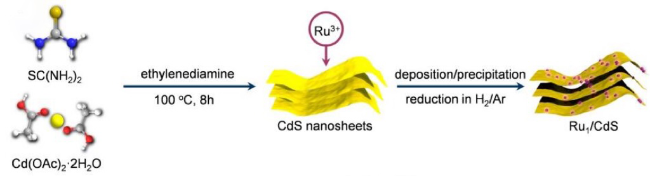
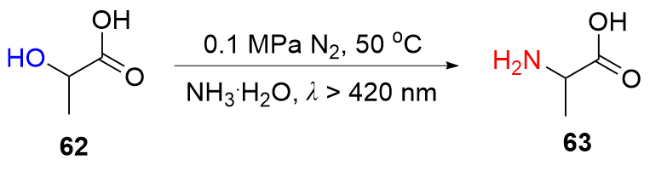
表2 Ru SAs/N-C和Ru纳米簇/C对喹啉选择性加氢的催化性能Table 2 Catalytic performance of Ru SAs/N-C and Ru nano- clusters/C for the selective hydrogenation of quinoline |
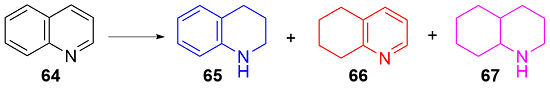
| Entry | Catalysts | Conv./% | Selec./% | ||
|---|---|---|---|---|---|
| 65 | 66 | 67 | |||
| 1 | Ru SAs/N-C | >99 | >99 | <1 | 0 |
| 2 | Ru SAs/C | >99 | 79 | 21 | 0 |
9 单原子光催化氧化

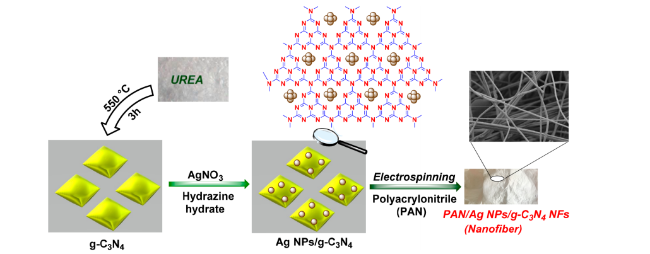
图14 PAN/Ag纳米和g-C3N4的合成示意图Figure 14 Synthesis diagram of PAN/Ag NPs/g-C3N4 NFs Copyright 2019 American Chemical Society. |
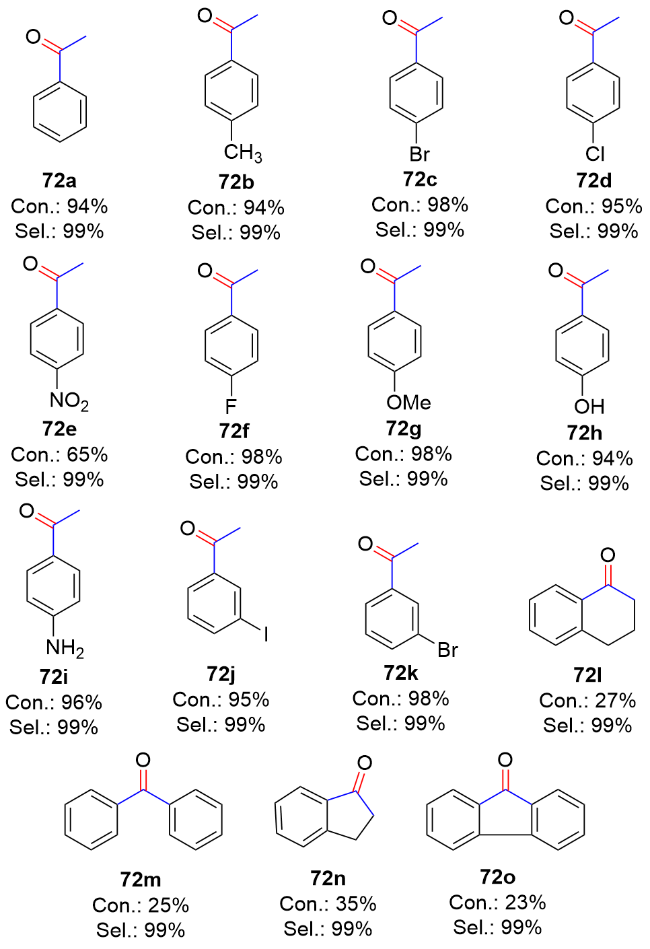
表3 PAN/Ag NPs/g-C3N4催化烯烃的选择性转化aTable 3 Selective conversion of olefins catalyzed by PAN/Ag NPs/g-C3N4 |
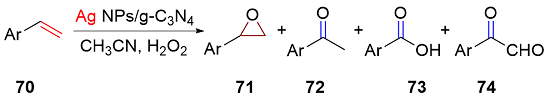
| Entry | Substrate | Conversion/% | Selectivity/% | |||
|---|---|---|---|---|---|---|
| 71 | 72 | 73 | 74 | |||
| 1 | 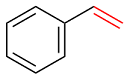 | 98 | 60 | 32 | 1 | 7 |
| 2 | 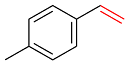 | 98 | 65 | 25 | — | 10 |
| 3 | 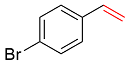 | 97 | 62 | 34 | — | 4 |
| 4 | 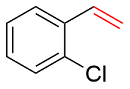 | 81 | 54 | 40 | 2 | 4 |
| 5 | 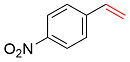 | 72 | 50 | 35 | 5 | 10 |
| 6 | 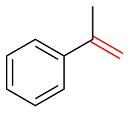 | 65 | 45 | — | — | — |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}