


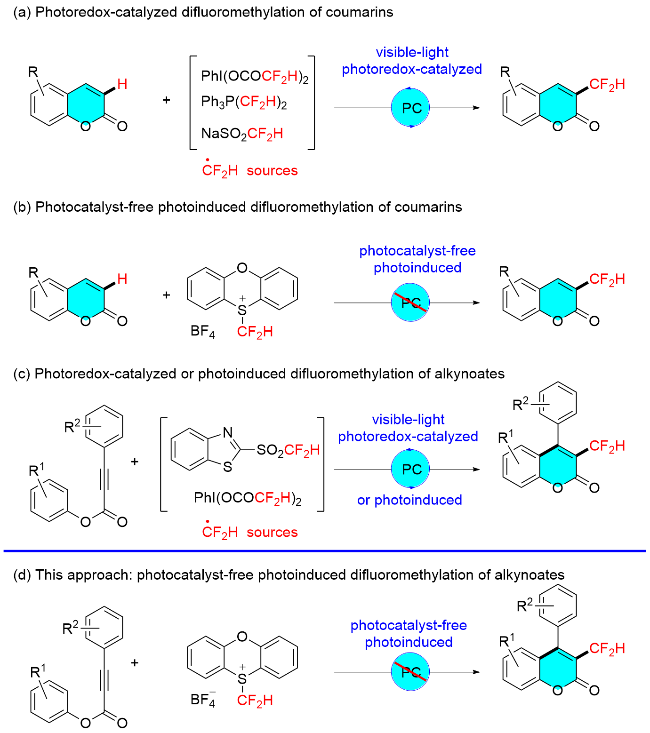
1 Introduction
Difluoromethyl (CF2H) is a marvelous and unique F- bearing functional group. It can serve as the more lipophilic hydrogen bond donors and bioisosteres of alcohols, thiols and amines,[1] as well as dramatically changing physicochemical and biological properties of parent molecules like other F-containing compounds.[1f,2] Thereby, incorporating CF2H into biologically active molecules or natural products will endow their improved bioactivities, lipophilicity, membrane permeability, metabolic stability, bioavailability and so on, resulting in CF2H-containing molecules widespread applications in pharmaceuticals,[1a,2a,3] agrochemicals[4] and materials.[5] Consequently, plenty of research effort goes into the development of synthetic methods for difluoromethylated functional molecules over the past two decades, particularly various structurally diverse CF2H- containing heterocycles due to their vital roles in pharmaceuticals and agrochemicals.[6] On the other hand, coumarins, including broad range of natural products and enormous amount of artificially synthetic derivatives, represent a large class of quite important benzopyrones containing π-π conjugated system with rich electron and good charge- transport properties. Their rigid fused ring structure endues coumarin-based derivatives great potential applications in pharmaceuticals, materials and supramolecular chemistry. Specially, coumarins have attracted considerable attention in new drug innovation and discovery due to a wide variety of pharmacological activities, such as anticoagulant, antineurodegenerative, anticancer, antioxidative, antibacterial, antifungal, antivirus, antiparasitic, antiinflammatory, antidiabetic and antidepressive actions etc.[7]
Given the reasons aforementioned, collection of CF2H- modified coumarin derivatives would great facilitate and accelerate finding of the lead compounds and new drug candidates. As a result, the synthesis of difluoromethylated coumarins is becoming of significant interest to synthetic chemistry and medicinal chemistry, and some examples were reported over the past few years. Recent years, difluoro- methyl radical chemistry has gained rapid advance, difluo- romethyl radical derived from various difluoromethyl sources via photoredox or electroredox strategies,[8] then leading to further various difluoromethyl radical transformation reactions. Thereby, difluoromethylated coumarins are efficiently accessible by dint of difluoromethyl radical chemistry. One synthetic strategy is the direct radical di- fluoromethylation of coumarins.[9-11] Deng, Wu, and Hu reported photoredox-catalyzed direct difluoromethylation of coumarins using CF2HSO2Na,[8] PhI(OCOCF2H)2,[10] and Ph3P(CF2H)2[11] as the difluoromethyl radical reagents via visible-light irradiation in the presence of appropriate photocatalyst, respectively (Scheme 1, a). Additionally, difluoro- methyl phenoxathiinium salt (PT- ), a shelf- stable and versatile difluoromethylating reagent developed recently by our group, is also a suitable reagent for the direct difluoromethylation of coumarins.[12] Notably, different of previous protocol, difluoromethyl phenoxathiinium salt facilitated this transformation in a manner of photocatalyst-free visible-light promotion (Scheme 1, b). Notably, radical triggered addition/cyclization cascade reaction of aryl alkynoates is an efficient pathway to access structurally-diverse 3-functionalized coumarins, and has been well documented over the past few years.[13] Therefore, as an alternative strategy, the radical difluoromethylation/cycli- zation cascade reaction of alkynoates is considered as a useful method for the preparation of difluoromethylated coumarins (Scheme 1, c).[14-15] In 2017, Fu and coworkers[14] described the photoredox-catalyzed difluoromethylation of alkynoates to access 3-CF2H-coumarins with ArSO2CF2H. Late on, Lu group[15] also achieved visible-light- induced radical difluoromethylation of alkynoates using PhI(OCOCF2H)2 as difluoromethyl radical precursor. Despite a few protocols for radical difluoromethylation/cycli- zation cascade reaction of alkynoates were achieved previously, most of them required the photocatalysts, especially the costly Ir- or Ru-based photosensitizers. Therefore, developing more concise, greener, milder and general approach to conveniently access CF2H-modified coumarin derivatives is still great desirable. Our difluoromethyl phenoxathiinium salt proved to be a good difluoromethyl radical reagent, which is capable of efficiently generating difluoromethyl radical under visible-light irradiation without externally adding photocatalyst, further resulting in various radical transformations. Herein, we report an elegant protocol for difluoromethylation of alkynoates with difluoromethyl phenoxa thiinium salt enabled by photocatalyst-free photo-mediation (Scheme 1, d).
2 Results and discussion
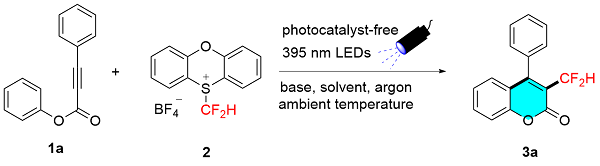
Our investigation commenced with radical difluoromethylation/cyclization of alkynoate 1a. As shown in Table 1, difluoromethyl phenoxathiinium salt 2 (2.5 equiv.) smoothly generated difluoromethyl radical species under 40 W 395 nm light irradiation in the absence of photosensitizer at ambient temperature, facilitating radical difluoromethylation/cyclization of alkynoate 1a (1.0 equiv.) with KH2PO4 (2.5 equiv.) as base to afford the desired 3-CF2H-coumarin 3a in 47% isolated yield (Table 1, Entry 1). Inspired by this result, various bases were further screened. Strong base LiOH gave the similar comparable yield of 45% (Table 1, Entry 2), but desired transformation was thoroughly inhibited using stronger t-BuOLi (Table 1, Entry 3). Delightedly, weaker base Li2CO3 significantly improved this transformation to increase yield to 78% (Table 1, Entry 4). Next, the effect of solvent was examined. Use of dichloromethane (DCM) and dichloroethane (DCE) also achieved good yields of 67% and 70%, respectively (Table 1, Entries 5, 6). However, EtOAc was less efficient resulting in decreased 43% yield (Table 1, Entry 7). The yield was further improved to 84% when 3.0 equiv. of reagent 2 was used (Table 1, Entry 8), while decreasing base to 2.0 equiv. resulted in less effective affording a lower yield of 60% (Table 1, Entry 9). Finally, the reaction time was surveyed, neither prolonging nor shortening time worked well (Table 1, Entries 10, 11). Longer time irradiation may cause decomposition of desired product, thus resulting in lower yield (Table 1, Entry 10).
Table 1 Optimization of reaction conditionsa |
| Entry | Base | Solvent | Time/h | Yield/% |
|---|---|---|---|---|
| 1 | KH2PO4 | CH3CN | 8 | 47 |
| 2 | LiOH | CH3CN | 8 | 45 |
| 3 | t-BuOLi | CH3CN | 8 | NR |
| 4 | Li2CO3 | CH3CN | 8 | 78 |
| 5 | Li2CO3 | DCM | 8 | 67 |
| 6 | Li2CO3 | DCE | 8 | 70 |
| 7 | Li2CO3 | EtOAc | 8 | 43 |
| 8b | Li2CO3 | CH3CN | 8 | 84 |
| 9c | Li2CO3 | CH3CN | 8 | 60 |
| 10b | Li2CO3 | CH3CN | 12 | 45 |
| 11b | Li2CO3 | CH3CN | 5 | 42 |
a Reaction conditions: 1a (0.1 mmol), 2 (0.25 mmol), base (0.25 mmol), solvent (2.0 mL), 395 nm (40 W), ambient temperature, under argon atmosphere. Isolated yields. b 3.0 equiv. of base was used. c 2.0 equiv. of base was used. |
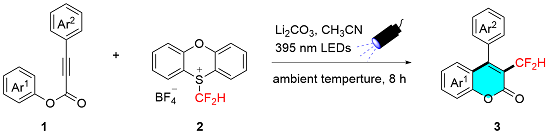
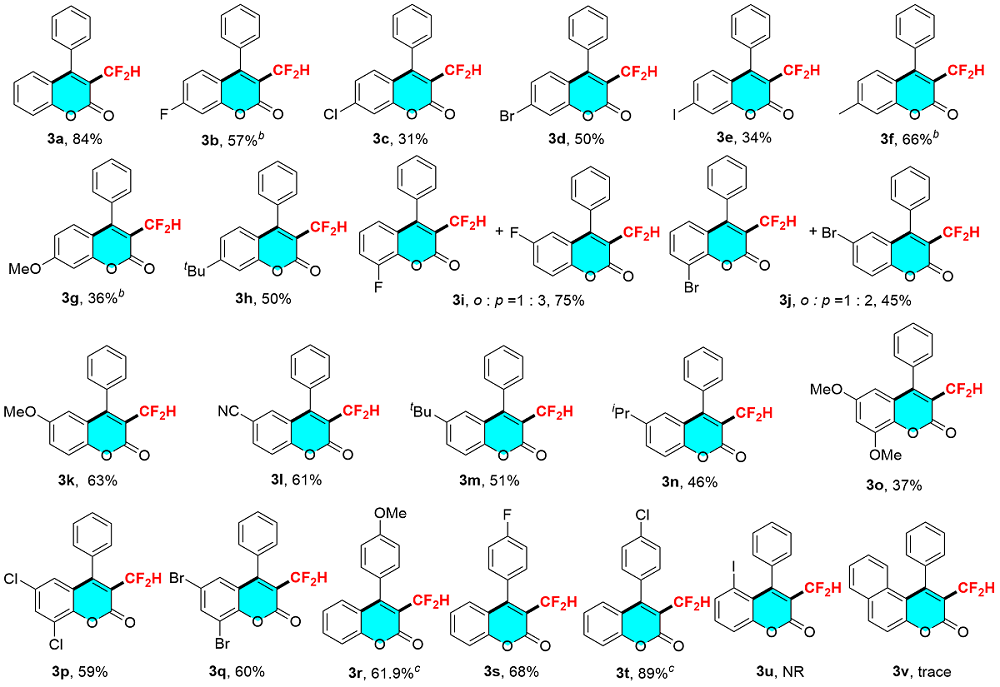
To demonstrate the generality and potential applications of this approach, the reaction scope of alkynoates with difluoromethylating reagent 2 was explored under the optimized reaction conditions obtained above (Table, Entry 8). As shown in Table 2, a broad range of alkynoates smoothly underwent difluoromethylation/cyclization reaction to give their corresponding 3-CF2H-coumarin in moderate to good yields. Firstly, the influences of substituents on Ar1 were investigated. para- and meta-substituted phenyl 3-phenyl- propiolates were suitable substrates for this transformation to deliver desired difluoromethylated coumarins in moderate to good yields, regardless of the position or electronic properties of substituents (3a~3n), whereas these substrates bearing ortho-substituent did not work or showed extremely low reactive (3u~3v). It should be noted that meta-F and Br-phenyl alkynoates 1i and 1j gave a mixture of ortho- and para-substituted coumarins 3i (o∶p=1∶3) and 3j (o∶p=1∶2), respectively. Particularly, synthetically useful halogens including fluorine, bromine, chlorine and iodine were compatible with this protocol to provide desired coumarins in moderate to good yields (3b~3e, 3i, 3j, 3p, 3q). Electron-donating group alkyl and alkoxy- substituted substrates were well tolerated to achieve good yields (3f~3h, 3k and 3m~3o). Moreover, di-substituted phenyl alkynoates also worked well to afford 3o, 3p and 3q in 37%, 59% and 60% yields, respectively. Longer reaction time of 12 h is necessary to obtain better results for cases 3b, 3f and 3g, while 3r and 3t accomplished higher yields within 5 h, which may be due to the decomposition under a long period of light irradiation. Notably, the properties of functional groups on Ar2 have almost no influence to the reaction, and desired difluoromethylation/cyclization pro-ceed with high reactivities regardless of whether electron- withdrawing and electron-donating groups were included, affording 3r, 3s and 3t in 61.9%, 68% and 89% yields, respectively.
Table 2 Substrate scopea |
 |
a Standard reaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2 (0.6 mmol, 3.0 equiv.), base (0.5 mmol, 2.5 equiv.), solvent (4.0 mL), 395 nm (40 W), ambient temperature, under argon atmosphere, 8 h. Isolated yields. b Reaction time 12 h. c Reaction time 5 h. |
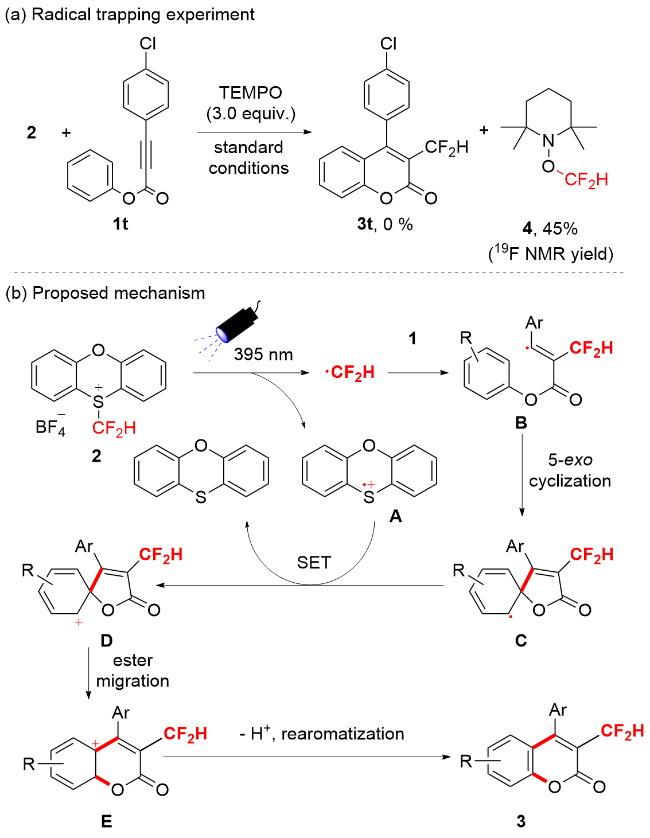
To gain detailed insight into the reaction mechanism, the radical trapping experiment was conducted. As shown in Scheme 2a, when radical scavenger 2,2,6,6-tetramethyl-1- piperidinyloxyl (TEMPO, 3.0 equiv.) was added to the standard reaction of mixture of 1t and 2, the desired transformation was completely depressed, and no desired product 3t was formed along with 45% 1-(difluoromethoxy)- 2,2,6,6-tetramethylpiperidine (4) generated from the trap of •CF2H radical with TEMPO. This result indicated that the reaction involved in •CF2H pathway. Based on the result of radical trapping experiment, previous literatures[14-15] and our own report,[12] a plausible difluoromethyl radical addition/5-exo radical cyclization cascade process is proposed in Scheme 2b. Initially, difluoromethylating reagent 2 happened homolysis under 395 nm light irradiation to deliver difluoromethyl radical (•CF2H) and radical cation A, followed by •CF2H adding to alkynoate 1 resulting in formation of intermediate radical B. Then B occurred intramolecular 5-exo cyclization to give spirocyclic radical intermediate C. Subsequently, single electron redox of C and radical cation A created spirocyclic cation species D along with generation of phenoxathiin. Finally, 1,2-ester migration of D formed cation E, followed by rearomatization by deprotonation to furnish desired difluoromethylated coumarin 3.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In conclusion, a photocatalyst-free photo-induced radical difluoromethylation of alkynoates by PT-$\mathrm{CF}_{2} \mathrm{H}^{+} \mathrm{BF}_{4}^{-}$ to facilitate ready synthesis of 3-CF2H-coumarins is reported. The reaction smoothly proceeded under mild photosensitizer-free conditions, and a broad scope of alkynoates were converted to desired 3-CF2H-coumarins in moderate to good yields with good functional groups tolerance. Additional, the rational difluoromethyl radical addition/spiro- cyclization/ester migration cascade process was proposed based on our previous research and related literature reports. Notably, this protocol also further extends the synthetic applications of the PT-$\mathrm{CF}_{2} \mathrm{H}^{+} \mathrm{BF}_{4}^{-}$ as a powerful and versatile difluoromethylating reagent.
4 Experimental section
4.1 General experimental information
Reagents and solvents were commercially available and were used without any further purification unless otherwise indicated. 1H NMR spectra were recorded on either a Bruker Ascend 400 MHz (400 MHz) spectrometer, or a Bruker Ascend 500 MHz (500 MHz) spectrometer at ambient temperature unless otherwise indicated. 13C NMR spectra were recorded on either a Bruker Ascend 500 MHz (126 MHz) spectrometer or a Bruker Ascend 600 MHz (151 MHz) spectrometer at ambient temperature and were proton decoupled. 19F NMR spectra were recorded on a Bruker Ascend 400 MHz (377 MHz) spectrometer or a Bruker Ascend 500 MHz (471 MHz) spectrometer at ambient temperature. ESI-MS analysis was performed in positive ionization mode on an Agilent 1260-Infinity LC/MSD resolution mass spectrometer. All high-resolution mass spectra were obtained on a Thermo Scientific Q-Exactive (HR/AM) Orbitrap mass spectrometer. Reactions were monitored by thin-layer chromatography (TLC) (detection with UV light). The products were purified by silica gel (300~400 mesh) chromatography. Visible light irradiation was performed by UV LED lamps (λ=395 nm; 20W×2) for preparative scale. Regent 2 was synthesized based on reported procedure and analytical data are in agreement with those reported in the literature.[12]
4.2 Synthesis of alkynaotats 1
3,5-Dichlorophenyl 3-phenylpropiolate (1p): Purified by silica gel chromatography (petrol ether/EtOAc, V∶V=50∶1) as a light yellow solid. m.p. 78.0~79.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.67 (d, J=6.9 Hz, 2H), 7.54 (t, J=7.6 Hz, 1H), 7.45 (t, J=7.5 Hz, 2H), 7.32 (s, 1H), 7.18 (d, J=1.9 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ: 151.28, 150.65, 135.44, 133.32, 131.39, 128.77, 126.86, 120.74, 118.90, 89.82, 79.60; HRMS (ESI) calcd for C15H8Cl2O2 [M+H]+ 291.1243, found 291.1270.
3,5-Dibromophenyl 3-phenylpropiolate (1q): Purified by silica gel chromatography ((petrol ether/EtOAc, V∶V=50∶1) as a white solid. m.p. 95.5~98.0 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.69~7.65 (m, 2H), 7.62 (t, J=1.6 Hz, 1H), 7.54 (t, J=7.5 Hz, 1H), 7.45 (t, J=7.4 Hz, 2H), 7.38 (d, J=1.6 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ: 151.32, 150.74, 133.35, 132.31, 131.41, 128.80, 124.01, 122.96, 118.91, 89.88, 79.59; HRMS (ESI) calcd for C15H8Br2O2 [M+H]+ 380.0377, found 380.0350.
4.3 General procedure for photoinduced difluoromethylation/cyclization cascade reaction
To a 10 mL Schlenk tube equipped with a magnetic stir bar were added alkynoate 1 (0.2 mmol, 1.0 equiv.), PT-$\mathrm{CF}_{2} \mathrm{H}^{+} \mathrm{BF}_{4}^{-}$ 2 (0.6 mmol, 202.8 mg, 3.0 equiv.) and Li2CO3 (0.5 mmol, 36.95 mg, 2.5 equiv.). The flask was flushed with argon, followed by the addition of acetonitrile (4.0 mL). The tube was placed at a distance of 2~3 cm away from UV LED lamps (λ=395 nm, 20 W×2), then the reaction mixture was stirred under irradiation of UV LEDs for 8 h at ambient temperature. The mixture was evaporated in vacuo after reaction accomplished, and the residue was purified by flash column chromatography on silica gel (petrol ether/EtOAc, V∶V=12∶1) to give desired product 3. All known products 3a~3o and 3r~3t were prepared according to the general procedure, and analytical data are in agreement with those reported in the previous literatures.[12-13]
3-(Difluoromethyl)-4-phenyl-2H-chromen-2-one (3a): Yellow solid (45.7 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ: 7.63 (t, J=8.6 Hz, 1H), 7.60~7.53 (m, 3H), 7.43 (d, J=8.4 Hz, 1H), 7.40~7.31 (m, 2H), 7.23 (t, J=8.2 Hz, 1H), 7.13 (d, J=8.1 Hz, 1H), 6.55 (t, J=53.3 Hz, 1H); 19F NMR (377 MHz, CDCl3) δ: -114.79 (d, J=52.7 Hz, 2F).
3-(Difluoromethyl)-6-fluoro-4-phenyl-2H-chromen-2-one (3b): Yellow solid (26.1 mg, 45% yield). 1H NMR (400 MHz, CDCl3) δ: 7.88~7.50 (m, 3H), 7.44~7.29 (m, 2H), 7.18~ 7.09 (m, 2H), 6.96 (td, J=8.5, 2.6 Hz, 1H), 6.51 (t, J=53.2 Hz, 1H); 19F NMR (377 MHz, CDCl3) δ: -102.34 (d, J=6.9 Hz), -114.74 (d, J=52.7 Hz, 2F).
3-(Difluoromethyl)-7-chloro-4-phenyl-2H-chromen-2-one (3c): Colorless oil (9.4 mg, 31% yield). 1H NMR (400 MHz, CDCl3) δ: 7.64~7.55 (m, 3H), 7.45 (d, J=2.0 Hz, 1H), 7.36~7.31 (m, 2H), 7.20 (dd, J=8.6, 2.0 Hz, 1H), 7.06 (d, J=8.6 Hz, 1H), 6.53 (t, J=53.2 Hz, 1H); 19F NMR (377 MHz, CDCl3) δ: -114.80 (d, J=53.4 Hz, 2F).
3-(Difluoromethyl)-7-bromo-4-phenyl-2H-chromen-2-one (3d): Orange solid (17.9 mg, 51% yield). 1H NMR (500 MHz, CDCl3) δ: 7.61 (d, J=1.8 Hz, 1H), 7.59 (dd, J=5.0, 1.7 Hz, 2H), 7.35~7.32 (m, 2H), 7.18~7.08 (m, 1H), 7.05~7.00 (m, 1H), 6.98 (d, J=8.5 Hz, 1H), 6.52 (t, J=53.2 Hz, 1H); 19F NMR (377 MHz, CDCl3) δ: -114.80 (d, J=53.4 Hz, 2F).
3-(Difluoromethyl)-7-iodo-4-phenyl-2H-chromen-2-one (3e): Reddish brown solid (34.3 mg, 34% yield). 1H NMR (500 MHz, CDCl3) δ: 7.81 (d, J=1.7 Hz, 1H), 7.63~7.53 (m, 4H), 7.35~7.30 (m, 2H), 6.81 (d, J=8.4 Hz, 1H), 6.52 (t, J=53.2 Hz, 1H); 19F NMR (471 MHz, CDCl3) δ: -114.84 (d, J=52.9 Hz, 2F).
3-(Difluoromethyl)-7-methyl-4-phenyl-2H-chromen-2-one(3f): Yellow solid (37.8 mg, 66% yield). 1H NMR (500 MHz, CDCl3) δ: 7.62~7.53 (m, 3H), 7.34 (dd, J=6.6, 3.1 Hz, 2H), 7.23 (s, 1H), 7.05~6.98 (m, 2H), 6.53 (t, J=53.3 Hz, 1H), 2.48 (s, 3H); 19F NMR (471 MHz, CDCl3) δ: -114.54 (d, J=53.8 Hz, 2F).
3-(Difluoromethyl)-7-methoxy-4-phenyl-2H-chromen-2- one (3g): Yellow solid (21.4 mg, 35.4% yield). 1H NMR (400 MHz, CDCl3) δ: 7.60~7.53 (m, 3H), 7.33 (dd, J=6.6, 2.9 Hz, 2H), 7.04~7.02 (m, 1H), 6.90 (d, J=2.5 Hz, 1H), 6.78 (dd, J=9.0, 2.5 Hz, 1H), 6.51 (t, J=53.4 Hz, 1H), 3.91 (s, 3H); 19F NMR (377 MHz, CDCl3) δ: -114.12 (d, J=53.4 Hz, 2F).
3-(Difluoromethyl)-7-(tert-butyl)-4-phenyl-2H-chromen-2-one (3h): Yellow solid (33 mg, 50% yield). 1H NMR (500 MHz, CDCl3) δ: 7.61~7.53 (m, 3H), 7.43 (d, J=1.8 Hz, 1H), 7.38 ~7.32 (m, 2H), 7.26 (dd, J=8.5, 1.9 Hz, 1H), 7.05 (d, J=8.5 Hz, 1H), 6.55 (t, J=53.3 Hz, 1H), 1.36 (s, 9H); 19F NMR (471 MHz, CDCl3) δ: -114.46 (d, J=52.9 Hz, 2F).
3-(Difluoromethyl)-6-fluoro-4-phenyl-2H-chromen-2-one (3i):[15] Yellow oil (43.5 mg, 75% yield). 1H NMR (500 MHz, CDCl3) δ: 7.61~7.57 (m, 4H), 7.43 (d, J=3.8 Hz, 0.3H), 7.43~7.40 (m, 1H), 7.39~7.31 (m, 4H), 6.90 (d, J=8.2 Hz, 0.3H), 6.80 (dd, J=8.8, 3.0 Hz, 1H), 6.55 (t, J=53.2 Hz, 1H), 6.54 (t, J=53.1 Hz, 0.3H); 19F NMR (471 MHz, CDCl3) δ: -114.96 (d, J=56.5Hz, 2F), -115.79~ -115.84 (m, 1F).
3-(Difluoromethyl)-6-bromo-4-phenyl-2H-chromen-2-one (3j):[15] Yellow oil (31.4 mg, 45% yield). 1H NMR (500 MHz, CDCl3) δ: 7.85 (dd, J=7.5, 1.8 Hz, 0.5H), 7.71 (dd, J=8.8, 2.3 Hz, 1H), 7.63~7.55 (m, 4.5H), 7.37~7.30 (m, 4H), 7.22 (d, J=2.3 Hz, 1H), 7.12~7.06 (m, 1H), 6.54 (t, J=53.2 Hz, 0.5H), 6.51 (t, J=53.2 Hz, 1H); 19F NMR (471 MHz, CDCl3) δ: -114.89 (d, J=53.8 Hz), -115.02 (d, J=52.9 Hz, 2F)
3-(Difluoromethyl)-6-methoxy-4-phenyl-2H-chromen-2-one (3k): Yellow solid (38.1 mg, 63% yield). 1H NMR (400 MHz, CDCl3) δ: 7.60~7.55 (m, 3H), 7.37 (d, J=9.1 Hz, 2H), 7.20 (dd, J=9.1, 2.9 Hz, 1H), 6.68~6.41 (m, 2H), 3.69 (s, 3H); 19F NMR (377 MHz, CDCl3) δ: -114.72 (d, J=53.4 Hz, 2F).
3-(Difluoromethyl)-2-oxo-4-phenyl-2H-chromene-6-carbonitrile (3l): Reddish brown solid (36.2 mg, 61% yield). 1H NMR (500 MHz, CDCl3) δ: 7.91 (dd, J=7.6, 1.8 Hz, 1H), 7.61 (dd, J=5.0, 2.1 Hz, 3H), 7.39~7.31 (m, 4H), 6.53 (t, J=52.9 Hz, 1H); 19F NMR (471 MHz, CDCl3) δ: -115.08 (d, J=52.9 Hz, 2F).
3-(Difluoromethyl)-6-(tert-butyl)-4-phenyl-2H-chromen-2-one (3m): Yellow solid (33.7 mg, 51% yield). 1H NMR (500 MHz, CDCl3) δ: 7.67 (dd, J=8.7, 2.4 Hz, 1H), 7.62~7.55 (m, 3H), 7.40~7.33 (m, 3H), 7.08 (d, J=2.4 Hz, 1H), 6.55 (t, J=53.3 Hz, 1H), 1.21 (s, 9H); 19F NMR (471 MHz, CDCl3) δ: -114.64 (d, J=53.8 Hz, 2F).
3-(Difluoromethyl)-6-isopropyl-4-phenyl-2H-chromen-2-one (3n): Yellow solid (28.9 mg, 46% yield). 1H NMR (600 MHz, CDCl3) δ: 7.65~7.55 (m, 3H), 7.39~7.36 (m, 2H), 7.16~7.12 (m, 1H), 7.06~7.03 (m, 1H), 6.55 (t, J=53.3 Hz, 1H), 2.87 (p, J=6.9 Hz, 1H), 1.18 (d, J=6.9 Hz, 6H); 19F NMR (471 MHz, CDCl3) δ: -115.24 (d, J=53.8 Hz, 2F).
3-(Difluoromethyl)-6,8-dimethoxy-4-phenyl-2H-chro-men-2-one (3o): Yellow solid (24.7 mg, 37.2% yield). 1H NMR (500 MHz, CDCl3) δ: 7.60~7.46 (m, 3H), 7.38~7.29 (m, 2H), 6.76 (d, J=2.6 Hz, 1H), 6.55 (t, J=53.3 Hz, 1H), 6.04 (d, J=2.7 Hz, 1H), 3.81 (d, J=170.9 Hz, 6H); 19F NMR (471 MHz, CDCl3) δ: -114.63 (d, J=52.9 Hz, 2F).
3-(Difluoromethyl)-6,8-dichloro-4-phenyl-2H-chromen-2-one (3p): Yellow solid (39.9 mg, 59% yield), m.p. 157~160 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.68 (d, J=2.4 Hz, 1H), 7.61 (dd, J=5.0, 2.1 Hz, 3H), 7.34~7.31 (m, 2H), 6.98 (d, J=2.4 Hz, 1H), 6.52 (t, J=53.0 Hz, 1H); 13C NMR (151 MHz, Chloroform-d) δ: 156.19, 155.06, 148.08, 133.34, 130.70, 130.32, 129.89, 129.12, 128.27, 126.44, 123.20, 121.66, 119.52 (t, J=22.5 Hz), 111.20 (t, J=240.0 Hz); 19F NMR (471 MHz, CDCl3) δ: -115.14 (d, J=52.9 Hz, 2F); HRMS (ESI) calcd for C16H8Cl2F2O2 [M+H]+ 341.1369, found 341.1348.
3-(Difluoromethyl)-6,8-dibromo-4-phenyl-2H-chromen-2-one (3q): Yellow solid (51.3 mg, 60% yield), m.p. 173~176 ℃; 1H NMR (600 MHz, Chloroform-d) δ: 7.96 (d, J=2.2 Hz, 1H), 7.63~7.57 (m, 3H), 7.32~7.28 (m, 2H), 7.14 (d, J=2.2 Hz, 1H), 6.49 (t, J=53.0 Hz, 1H); 13C NMR (151 MHz, Chloroform-d) δ: 156.22, 154.94, 149.48, 138.91, 130.57, 130.29, 130.09, 129.09, 128.23, 121.98, 119.46 (d, J=22.7 Hz), 117.29, 111.81, 111.12 (t, J=240.0 Hz); 19F NMR (471 MHz, CDCl3) δ: -115.11 (d, J=52.9 Hz, 2F); HRMS (ESI) calcd for C16H8Br2F2O2 [M+H]+ 430.0457, found 430.0428.
3-(Difluoromethyl)-4-(4-methoxyphenyl)-2H-chromen-2-one (3r): Yellow solid (37.4 mg, 61.9% yield). 1H NMR (500 MHz, CDCl3) δ: 7.65~7.59 (m, 1H), 7.42 (d, J=8.2 Hz, 1H), 7.30~7.27 (m, 2H), 7.23 (qd, J=8.1, 1.8 Hz, 2H), 7.09 (d, J=8.9 Hz, 2H), 6.56 (t, J=53.3 Hz, 1H), 3.93 (s, 3H); 19F NMR (471 MHz, CDCl3) δ: -114.76 (d, J=52.9 Hz, 2F).
3-(Difluoromethyl)-4-(4-fluorophenyl)-2H-chromen-2-one (3s): Yellow solid (39.4 mg, 68% yield). 1H NMR (500 MHz, CDCl3) δ: 7.64 (t, J=8.7 Hz, 1H), 7.43 (d, J=7.2 Hz, 1H), 7.35 (dd, J=8.9, 5.2 Hz, 2H), 7.30~7.23 (m, 3H), 7.12 (d, J=1.7 Hz, 1H), 6.62 (t, J=53.3 Hz, 1H); 19F NMR (471 MHz, CDCl3) δ: -110.60, -113.96 (d, J=52.9 Hz, 2F).
3-(Difluoromethyl)-4-(4-chlorophenyl)-2H-chromen-2-One (3t): brown solid (54.7 mg, 89.2 % yield). 1H NMR (500 MHz, CDCl3) δ: 7.64 (t, J=8.6 Hz, 1H), 7.56 (d, J=8.4 Hz, 2H), 7.43 (d, J=8.4 Hz, 1H), 7.30 (d, J=8.4 Hz, 2H), 7.25 (t, J=8.2 Hz, 1H), 7.12 (dd, J=6.7, 1.8 Hz, 1H), 6.64 (t, J=53.3 Hz, 1H); 19F NMR (471 MHz, CDCl3) δ: -113.72 (d, J=52.9 Hz, 2F).
4.4 Mechanistic study: radical trapping experiment
To a 10 mL Schlenk tube equipped with a magnetic stir bar were added alkynoate 1t (0.2 mmol, 1.0 equiv.), PT-$\mathrm{CF}_{2} \mathrm{H}^{+} \mathrm{BF}_{4}^{-}$ 2 (0.6 mmol, 202.8 mg, 3.0 equiv.), TEMPO (0.6 mmol, 96.3 mg, 3.0 equiv.) and Li2CO3 (0.5 mmol, 36.95 mg, 2.5 equiv.). The flask was flushed with argon, followed by the addition of acetonitrile (4.0 mL). The tube was placed at a distance of 2~3 cm away from UV LED lamps (λ=395 nm, 20 W×2), then the reaction mixture was stirred under irradiation of UV LEDs for 8 h at ambient temperature. The mixture was evaporated in vacuo and the residue was subjected to 19F NMR. The result indicated that no 3t was formed, and •CF2H trapping product 1-(difluoro- methoxy)-2,2,6,6-tetramethylpiperidine (4) was detected (45% 19F NMR yield).
Acknowledgement We thank the Instrumental Analysis Center of Shenzhen University for its analytical work.
Supporting Information General procedure for the pre- paration of products 3, reaction apparatus, characterization data for 3-phenylpropargyl ester 1, characterization data for difluoromethylated coumarins 3 and NMR spectra for new compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Zhao, C.)