


1 Introduction
The ketone group is a versatile and useful structural motif that is frequently distributed in a myriad of naturally occurring products and synthetically bioactive compounds.[1] Moreover, ketones could act as fundamental feedstocks in the preparation of natural products, active pharmaceutical ingredients, and other important derivatives in organic synthesis because of their synthetic accessibility and intrinsic ability to be converted to other diverse functional groups.[2] Consequently, chemists have made long-standing efforts to establish various efficient synthetic tactics for the synthesis of functionalized ketones.[3] Important methods for the synthesis of ketones include the oxidation of alcohols,[4] electrophilic acylation (acylation of carbon-centered nucleophiles),[5] the addition of acyl radicals across unsaturated systems,[6] and the acylation of carbon-centered radicals (radical acylation)[7] (Scheme 1a). Among them, the addition of acyl radicals across unsaturated carbon-carbon bonds has become a primary and powerful shortcut for the fabrication of ketones, benefiting from controllable radical-transfer direction, rich sources of acyl radicals, and availability of the reaction.[8] Specifically, a variety of acyl radical precursors, including aroyl chlorides,[9] aryl anhydrides,[10] acylsilanes,[11] 2-pyri-dylthioesters,[12] carboxylic acids,[13] aldehydes,[14] α-keto acids,[15] α-hydroxylketones,[16] 4-acyl-1,4-dihydropyri-dines[17] and acyl oxime esters,[18] have been exploited, enabling single electron transfer (SET) under visible-light photocatalysis to access acylated products via acyl radical species. Despite these achievements, the development of new and efficient approaches for the generation of functionalized ketones with structural diversity via acyl radical-triggered transformations is highly desirable.
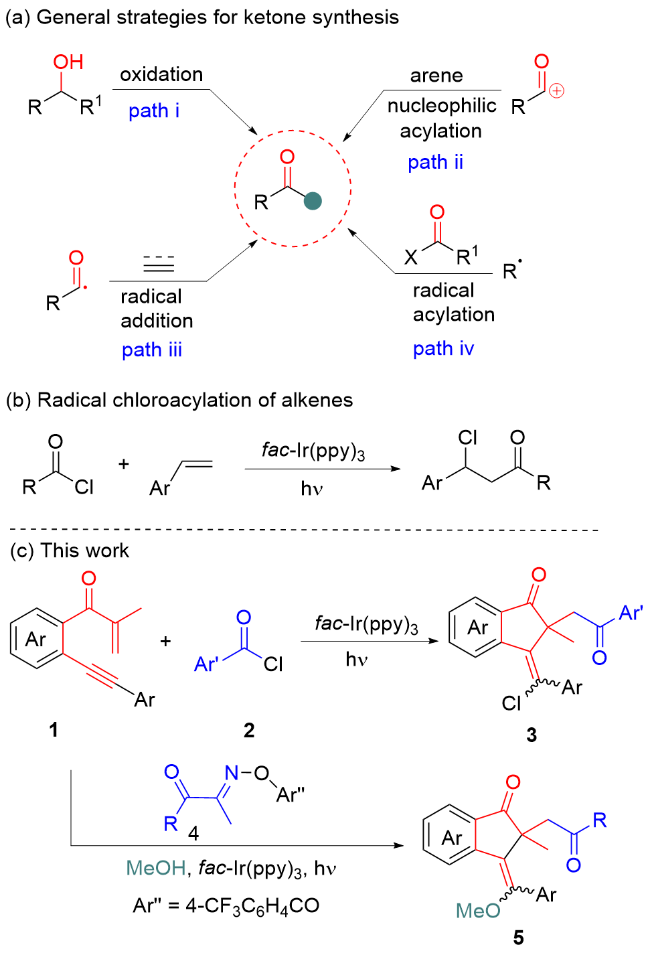
Visible-light-driven photoredox catalysis (VLPC) has become a flourishing and powerful tool for initiating various synthetic transformations under usually mild conditions by virtue of the impressive characteristics of visible light, including green, sustainability, and renewable energy sources.[19] Generally, photoredox catalysis can produce significant radical species with frequently unusual or unconventional reactivities via a single-electron transfer process, which are engaged in a remarkable variety of unusual chemical transformations to derive biologically important molecules that are difficult to obtain via thermal processes. Over the years, many state-of-the-art and seminal works have demonstrated the tremendous potential of visible light photoredox catalysis in organic synthesis.[20] Notably, Ngai,[21] Oh[22] and co-workers independently reported elegant photocatalytic acylation-functionalization of alkenes using aroyl chlorides as acyl radical precursors, providing a practical pathway for producing β-functionalized ketones. However, a survey of the literature reveals that there are no reports on the photoredox catalysis of 1,6-enynes with in situ generated acyl radical species to form acylated 1-indanones. To continue our interest in VLPC,[23] herein, we report a new and external oxidant-free annulative acylative difunctionalization of 1,6-enynes to synthesize 1- indanones via the photocatalysis of easily accessible substrates under very mild conditions. The use of aroyl chlorides as bifunctional reagents of chlorine and acyl sources enabled the chloroacylation of 1,6-enynes via an atom transfer radical cyclization (ATRC) approach, whereas annulative alkoxyacylation of 1,6-enynes can be realized by exploiting acyl oxime esters as acyl radical precursors and alcohols as alkoxylation reagents.
2 Results and discussion
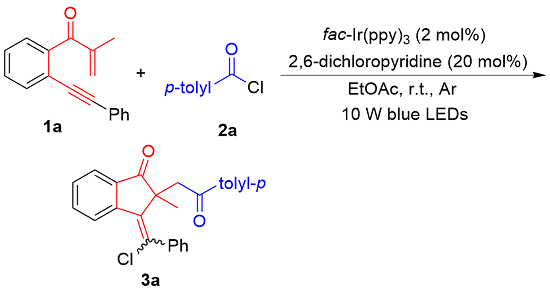
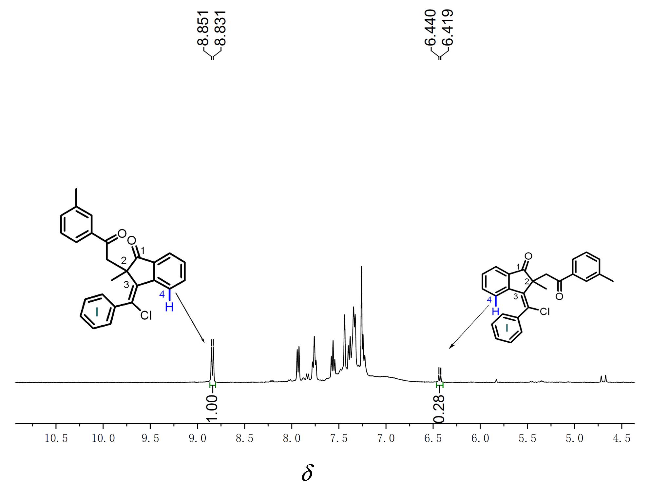
At the beginning of our study, the preformed 1,6-enyne 1a and p-tolyl chloride (2a) were selected as model substrates to screen the reaction conditions (Table 1), and the results are summarized in Table 1. The reaction proceeded readily in ethyl acetate (EtOAc) under argon conditions with the irradiation of 10 W blue LEDs by using fac-Ir(ppy)3 (2 mol%) as a photocatalyst and 2,6-dichloropyri-dine (20 mol%) as a base, and the expected 1-indanone 3a was provided in 85% yield with a >19∶1 E/Z ratio (Table 1, Entry 1). The E/Z ratio was determined through 1H NMR analysis and by analogy with 3b as a case study (Figure 1).[24] Decreasing or increasing the catalytic amount of fac-Ir(ppy)3 did not improve the yield of 3a (Entries 2, 3). Subsequent screening of several other organic bases, such as 2,4,6-collidine, 2,6-lutidine and 4- dimethylaminopyridine (DMAP) (Entries 4~6), revealed that none of them improved in the yield. The decrease in the loading of 2,6-dichloropyridine resulted in a marked decrease in the yield of 3a (56%, Entry 7). The reaction did not proceed when ethyl acetate (EtOAc) was changed to tetrahydrofuran (THF) as the reaction medium (Entry 8). The reaction became messy under air conditions, and only a trace amount of 3a was detected (Entry 9), implying that O2 could seriously deteriorate this transformation. Exchanging fac-Ir(ppy)3 with Ru(bpy)3Cl2 completely suppressed the reaction process (Entry 10). No product 3a was observed without the fac-Ir(ppy)3 catalyst or light irradiation (Entries 11, 12), suggesting that both the photocatalyst and visible light are the keys to driving this transformation.
Table 1 Condition optimization for product 3aa |
| Entry | Variation of the established conditions | Yieldb/% |
|---|---|---|
| 1 | None | 85 |
| 2 | 1.0 mol% fac-Ir(ppy)3 | 77 |
| 3 | 5.0 mol% fac-Ir(ppy)3 | 85 |
| 4 | 2,4,6-Collidine (20 mol%) as the base | 30 |
| 5 | 2,6-Lutidine (20 mol%) as the base | 45 |
| 6 | DMAP (20 mol%) as the base | 0 |
| 7 | 2,6-Dichloropyridine (10 mol%) | 56 |
| 8 | THF instead of EtOAc | NR |
| 9 | In the air | Trace |
| 10 | Ru(bpy)3Cl2•6H2O instead of fac-Ir(ppy)3 | Trace |
| 11 | Without fac-Ir(ppy)3 | 0 |
| 12 | No light | 0 |
a Reaction conditions: 1a (0.2 mmol), 2a (2.0 equiv.), fac-Ir(ppy)3 (2 mol%), 10 W Blue LEDs, EtOAc (2.5 mL) under Ar condition at 25 ℃ for 24 h. b Isolated yield based on 1a. ppy=2-phenylpyridine; bpy=bipyridine, NR=no reaction. |
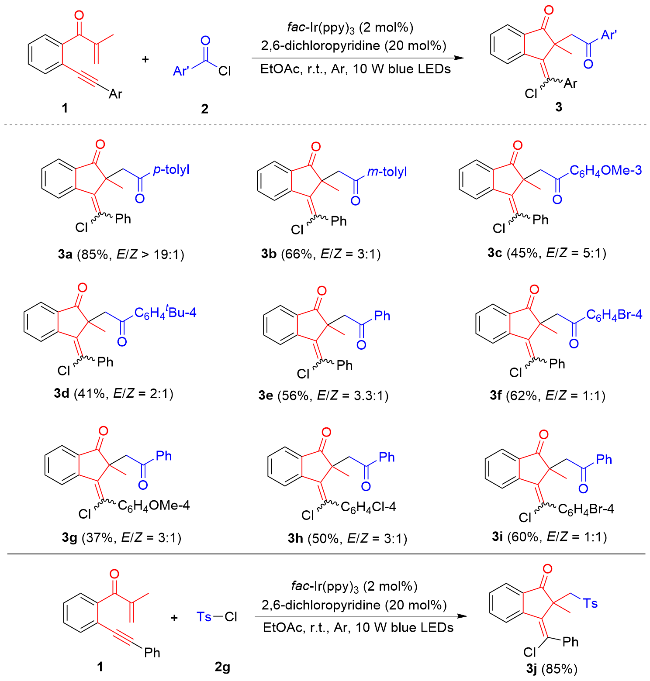
With these established reaction conditions in hand, the generality of photocatalytic annulative chloroacylation with respect to 1,6-enynes and aroyl chlorides was probed (Scheme 2). First, the scope of aroyl chlorides was evaluated, and several aroyl chlorides with different electronic properties at both aromatic rings were found to be compatible as illustrated in Scheme 2. Electronically rich (e.g. meta-methyl 2b, meta-methoxy 2c, para-tert-butyl 2d), neutral (H 2e) and poor (para-bromo 2f) groups at different positions (para and meta) in the arene ring were well accommodated, enabling visible-light-induced annulation to furnish products 3b~3f in 41%~66% yields and between 1∶1 and 5∶1 E/Z ratios. Similarly, 1,6-enyne bearing methoxy (1b), chloro (1c), or bromo (1d) group at the para-position in the aromatic ring directly bound to the alkyne unit (Ar) was workable for this catalytic protocol, delivering products 3g~3i in acceptable yields, albert with moderate E/Z ratios. Notably, swapping aroyl chloride with p-toluenesulfonyl chloride (TsCl) 2g enabled a completely stereoselective pathway to generate sulfonylated 1-inda- none 3j as a single E-stereoisomer in 85% yield.
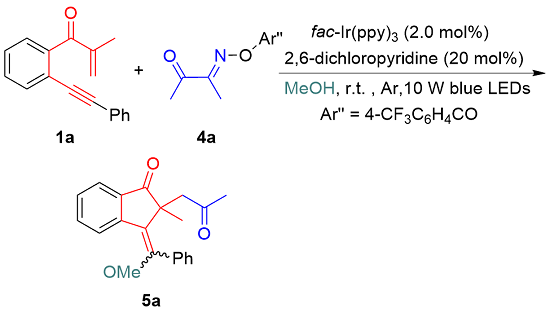
After achieving photocatalytic annulative chloroacylation of 1,6-enynes, we turn our attention to evaluate the feasibility of annulative alkoxyacylation of 1,6-enynes.[25] The reaction of 1,6-enyne 1a with benzoyl chloride was conducted in MeOH under standard conditions (Scheme 3). However, the reaction did not proceed through alkoxyacylation, probably because of the esterification between benzoyl chloride and MeOH in the presence of bases. To avoid esterification, we selected acyl oxime esters as acyl radical precursors for alkoxyacylation.[18] To our delight, the reaction of 1a with acyl oxime ester 4a proceeded smoothly along the direction of alkoxylation, and desired product 5a was obtained in 45% yield with a 6∶1 Z/E ratio (Scheme 3). After simple optimization of the reaction conditions, the yield of 5a was improved to 65% when the reaction worked in the presence of 1 mol% fac-Ir(ppy)3 without any base (Table 2, Entry 3). Changing other reaction parameters, including the solvent (e.g., EtOH), inorganic base (e.g., K2CO3 and Na2CO3), and photocatalyst (e.g., Eosin Y and Ru(bpy)3Cl2•6H2O) did not enhance the efficiency of this transformation (Entries 4~8). Moreover, a lower conversion of 1a into 5a was detected under air conditions (Table 2, Entry 9). No reactions occurred without fac-Ir(ppy)3 or light irradiation (Entries 10, 11).
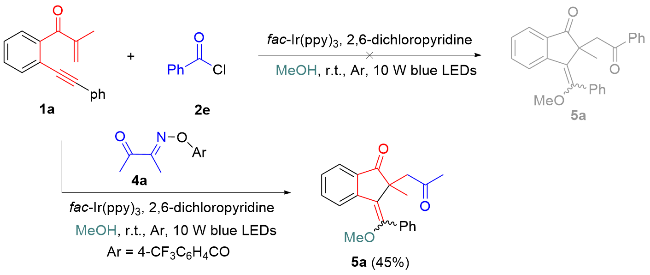
Table 2 Condition optimization for product 5aa |
| Entry | Variation of the established conditions | Yieldb/% |
|---|---|---|
| 1 | None | 45 |
| 2 | 1.0 mol% of fac-Ir(ppy)3 | 45 |
| 3 | 1.0 mol% of fac-Ir(ppy)3 without base | 65 |
| 4 | EtOH instead of MeOH | Trace |
| 5 | K2CO3 (2.0 equiv.) | 21 |
| 6 | Na2CO3 (2.0 equiv.) | 53 |
| 7 | Eosin Y instead of fac-Ir(ppy)3 | 32 |
| 8 | Ru(bpy)3Cl2•6H2O instead of fac-Ir(ppy)3 | Trace |
| 9 | In the air | 55 |
| 10 | Without fac-Ir(ppy)3 | 0 |
| 11 | No light | 0 |
a Reaction conditions: 1a (0.2 mmol), 4a (1.5 equiv.), fac-Ir(ppy)3 (1 mol%), 10 W Blue LEDs, MeOH (2.0 mL) under Ar conditions at 25 ℃ for 24 h. b Isolated yield based on 1a. ppy=2-phenylpyridine; bpy=bipyridine. |
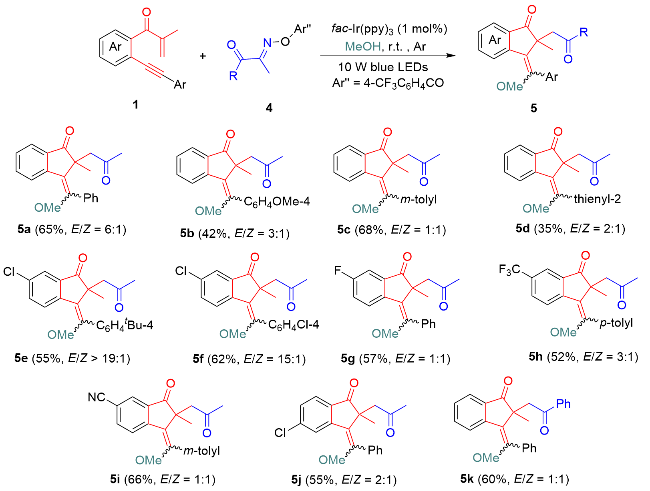
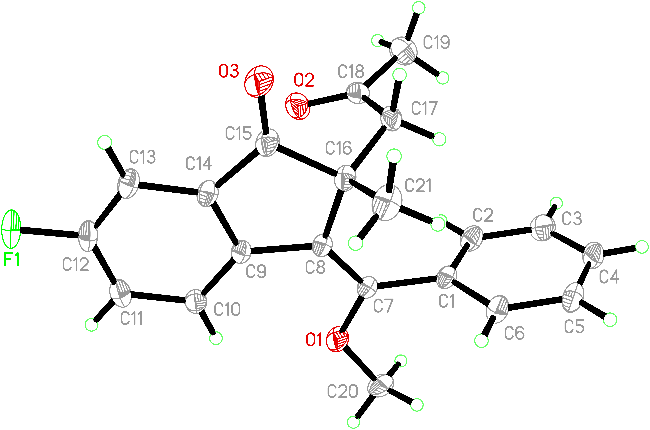
Following these acceptable conditions, we extended our studies to the scope of 1,6-enynes and acyl oxime esters for annulative alkoxyacylation (Scheme 4). Substituents inc-luding methoxy and methyl at different positions of the arylalkynyl (Ar) moiety were converted into corresponding products 5b and 5c in 42% and 68% yields, respectively. Notably, the 2-thienyl counterpart showed high reactivity, providing 2-thienyl-substituted 1-indanone 5d with inseparable stereoisomers in 35% yield. Next, various functional groups, such as chloro, fluoro, trifluoromethyl, and cyano, at the C4 or C5 position in the internal arene ring were proven to be applicable for this alkoxyacylation, as products 5e~5j with acceptable yields were afforded. Among these groups, two challenging cases with trifluoromethyl and cyano functionalities as strongly electronically deficient groups were effective for this catalytic protocol, demonstrating the good compatibility of this photocatalysis. Swapping the acetyl group with a benzoyl group in the oxime ester substrate led to the formation of product 5k with equal amounts of stereoisomers in 60% yield. Moreover, a scale-up reaction of 1a with 4a was conducted with a 1.0 mmol scale, furnishing product 5a in a slightly decreased 60% yield. The resulting 1-indanones were fully identified via NMR spectroscopy and HRMS data. Furthermore, the stereo-structure of 5g was confirmed by X-ray diffraction analysis (Figure 2, CCDC 2387944). Moreover, Z/E-configuration was confirmed by 1H NMR analysis (Figure 1). Using compound 3b as a case study, when exocyclic double bond is in Z-configuration, the proton at the C4 position of the 1-indenone ring is shielded by the benzene ring I, resulting in an upfield shift of its 1H NMR absorption to appear as a doublet at δ 6.43.[25] In contrast, when the exocyclic double bond adopts in E-configuration, there is no shielding effect between the proton at C4 position and the benzene ring I, thus remains unshifted and appears as a doublet at δ 8.84. In the case of compound 3b, E-isomer predominates as the major stereoisomer.
To elucidate this mechanism of photocatalysis, the reactions of 1a with 2a (or 4a) in the presence of 2,2,6,6-tetra- methyl-piperidine-N-oxyl (TEMPO, 2.0 equiv.) were carried out under standard conditions (Schemes 5a and 5b). As a result, this process was severely inhibited, and TEPMO-acyl adducts were detected by HRMS and GC-MS, respectively, indicating that an acyl radical process may be involved.

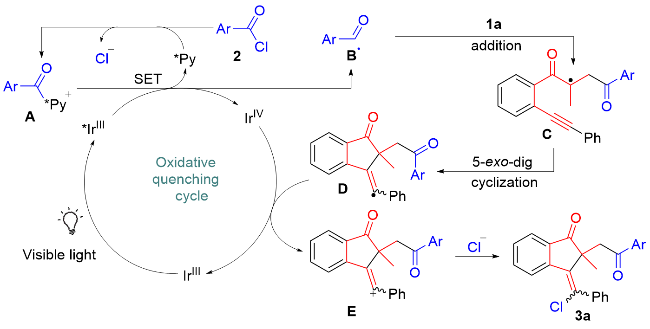
Combined with these observations and literature reports,[21,22,24,26] a reasonable reaction pathway for the formation of products 3 is proposed in Scheme 6. Upon exposure to visible light, the excited state of the *Ir(III) complex, which is rapidly generated from the normal Ir(III) complex, reduces N-acylpyridinium ion A, derived from aroyl chloride 2 and 2,6-dichloropyridine, via single electron transfer (SET) to generate acyl radical B,[27] together with Ir(IV) species and 2,6-dichloropyridine. Next, acyl radical B undergoes regioselective addition and 5-exo-dig- cyclization to produce alkenyl radical D. Intermediate D is oxidized by Ir(IV) species to yield alkenyl cation E and regenerate the Ir(III) complex for the next catalytic cycle. The former cation E is captured by the chloride anion to afford the final product 3a. The formation of 5a is similar to the above proposed pathway.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In summary, we have developed a new photocatalytic annulative acylative difunctionalization of 1,6-enynes under mild conditions, enabling stereoselective access to ac ylated 1-indanones with a quaternary center in moderate to good yields. An annulative chloroacylation of 1,6-enynes was realized by taking advantage of aroyl chlorides as bifunctional reagents of chlorine and acyl sources, whereas the three-component annulative alkoxyacylation of 1,6- enynes produced acylated 1-indanones with acyl oxime esters as acyl radical precursors and alcohols as the alkoxylation reagents. The current method provides a unified and operationally simple strategy for preparing 1-inda- nones with substitution diversity, featuring good functional group compatibility and a broad substrate scope. Further application of photocatalytic acylative functionalization for the forging of important cyclic structures is underway in our laboratory.
4 Experimental section
4.1 Materials and methods
1H NMR (13C NMR) spectra were measured on a Bruker DPX 400 MHz spectrometer in CDCl3 with chemical shift (δ) given relative to TMS as internal standard. HRMS (APCI and ESI) was determined by using microTOF-QII HRMS/MS instrument (BRUKER).
4.2 General procedure for the synthesis of products 3a and 5a
To a dry Schlenk reaction tube equipped with a magnetic stir bar, were added 1,6-enyne 1a (49.2 mg, 0.2 mmol), 4-methylbenzoyl chloride (2a, 61.6 mg, 0.4 mmol), and fac-Ir(ppy)3 (0.004 mmol, 2.6 mg). The Schlenk tube was closed with a septum, evacuated and refilled with Ar conditions. 2,6-Dichloropyridine (7.6 mg, 0.04 mmol) and dry EtOAc (2.5 mL) was added via syringe. The mixture was placed in LH LABWARE photosynthesis instrument by 10 W Blue-LED lamp and stirred at room temperature and monitored by thin-layer chromatography (TLC) for 24 h. The mixture was concentrated under reduced pressure and purified by column chromatography on silica gel (elution with petroleum ether to 30∶1 ether/EtOAc). to afford the desired product 3a (85% yield) as a yellow solid.
To a dry Schlenk reaction tube equipped with a magnetic stir bar, was added 1,6-enyne 1a (49.2 mg, 0.2 mmol), acyl oxime ester 4a (82 mg, 0.03 mmol), fac-Ir(ppy)3 (0.002 mmol, 1.3 mg, 1 mol%). The Schlenk tube was closed with a septum, evacuated and refilled with Ar conditions. MeOH (2.0 mL) was added via syringe. The mixture was placed in LH LABWARE photosynthesis instrument by 10W Blue-LED lamp and stirred at room temperature and monitored by TLC for 24 h. The mixture was concentrated under reduced pressure and purified by via column chromatography on silica gel (elution with petroleum ether to 10∶1 ether/EtOAc) to afford the desired product 5a (65% yield) as a yellow solid.
4.3 Scale-up reaction of 1a
To a dry Schlenk reaction tube equipped with a magnetic stir bar, were added 1,6-enyne 1a (246 mg, 1 mmol), acyl oxime ester 4a (410 mg, 0.15 mmol), and fac-Ir(ppy)3 (0.01 mmol, 6.5 mg, 1 mol%). The Schlenk tube was closed with a septum, evacuated and refilled with Ar. MeOH (4.0 mL) was added via syringe. The mixture was placed in LH LABWARE photosynthesis instrument by 10 W Blue-LED lamp and stirred at room temperature and monitored by TLC for 24 h. The mixture was concentrated under reduced pressure and purified by column chromatography on silica gel (elution with petroleum ether to 10∶1 ether/EtOAc) to afford the desired product 5a (192 mg, 60% yield) as a yellow solid.
4.4 Compound characterization
(E)-3-(Chloro(phenyl)methylene)-2-methyl-2-(2-oxo-2-(p-tolyl)ethyl)-2,3-dihydro-1H-inden-1-one (3a): White solid, 68.1 mg, 85% yield. m.p. 138~140 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.84 (d, J=8.0 Hz, 1H), 7.93 (d, J=7.6 Hz, 1H), 7.80~7.72 (m, 1H), 7.58~7.53 (m, 1H), 7.51 (d, J=7.6 Hz, 2H), 7.49~7.39 (m, 1H), 7.38~7.29 (m, 2H), 7.22~7.13 (m, 3H), 7.12~7.06 (m, 1H), 3.32 (d, J=17.2 Hz, 1H), 2.76 (d, J=18.8 Hz, 1H), 2.39 (s, 3H), 1.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.1, 196.1, 146.8, 144.1, 139.7, 138.6, 135.9, 134.6, 133.3, 129.5, 129.1 (d, J=9.1 Hz), 124.1, 122.7, 52.1, 46.0, 21.8. HRMS (ESI) calcd for C26H22ClO2 [M+H]+ 401.1308, found 401.1304.
(E)-3-(Chloro(phenyl)methylene)-2-methyl-2-(2-oxo-2-(m-tolyl)ethyl)-2,3-dihydro-1H-inden-1-one (3b, major): Yellow solid, 52.9 mg, 66% yield. m.p. 131~133 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.84 (d, J=8.0 Hz, 1H), 7.93 (d, J=7.6 Hz, 1H), 7.86~7.72 (m, 2H), 7.58~7.54 (m, 1H), 7.46 (d, J=17.6 Hz, 2H), 7.42~7.31 (m, 5H), 7.23 (d, J=7.2 Hz, 1H), 3.32 (d, J=18.8 Hz, 1H), 2.78 (d, J=18.8 Hz, 1H), 2.34 (s, 3H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.0, 196.6, 146.8, 139.7, 138.2, 134.6, 134.0, 129.5, 129.0, 128.7, 128.3, 127.5, 125.4, 124.1, 52.1, 46.1, 25.7, 21.4. HRMS (ESI) calcd for C26H22ClO2 [M+H]+ 401.1308, found 401.1313.
(E)-3-(Chloro(phenyl)methylene)-2-(2-(3-methoxyphen-yl)-2-oxoethyl)-2-methyl-2,3-dihydro-1H-inden-1-one (3c, major): Yellow solid, 37.9 mg, 45% yield. m.p. 140~142 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.85 (d, J=8.0 Hz, 1H), 7.94 (d, J=7.6 Hz, 1H), 7.81~7.74 (m, 1H), 7.59~7.54 (m, 1H), 7.49~7.41 (m, 1H), 7.38~7.32 (m, 2H), 7.28~7.21 (m, 3H), 7.18~7.03 (m, 3H), 3.78 (s, 3H), 3.34 (d, J=18.8 Hz, 1H), 2.77 (d, J=18.8 Hz, 1H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 205.9, 196.3, 159.7, 146.7, 139.6, 138.4, 137.0, 135.8, 134.7, 129.5, 129.3, 129.0, 127.5, 124.1, 120.9, 120.3, 111.6, 55.4, 52.1, 46.1, 25.6. HRMS (ESI) calcd for C26H22ClO3 [M+H]+ 417.1257, found 417.1266.
(E)-2-(2-(4-(tert-Butyl)phenyl)-2-oxoethyl)-3-(chloro-(phenyl)methylene)-2-methyl-2,3-dihydro-1H-inden-1-one (3d, major): Yellow solid, 36.3 mg, 41% yield. m.p. 135~137 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.84 (d, J=8.0 Hz, 1H), 7.99~7.85 (m, 2H), 7.79~7.73 (m, 1H), 7.59~7.51 (m, 3H), 7.50~7.42 (m, 2H), 7.37 (d, J=8.0 Hz, 4H), 3.32 (d, J=18.8 Hz, 1H), 2.78 (d, J=18.4 Hz, 1H), 1.33 (s, 9H), 1.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.0, 196.1, 157.0, 146.8, 139.7, 138.6, 135.9, 134.6, 134.0, 129.5, 128.2, 128.1, 125.6, 125.3, 124.1, 52.1, 46.1, 35.2, 31.2, 25.6, 21.8. HRMS (ESI) calcd for C29H28ClO2 [M+H]+ 443.1778, found 443.1780.
(E)-3-(Chloro(phenyl)methylene)-2-methyl-2-(2-oxo-2-phenylethyl)-2,3-dihydro-1H-inden-1-one (3e, major): Yellow solid, 43.3 mg, 56% yield. m.p. 142~144 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.85 (d, J=8.4 Hz, 1H), 7.95~7.91 (m, 1H), 7.89~7.68 (m, 2H), 7.61 (d, J=7.7 Hz, 2H), 7.60~7.49 (m, 3H), 7.47~7.41 (m, 1H), 7.39~7.30 (m, 4H), 3.35 (d, J=18.8 Hz, 1H), 2.76 (d, J=18.8 Hz, 1H), 1.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 205.9, 196.4, 146.7, 139.6, 138.5, 135.7, 134.6, 133.2, 129.5, 129.0, 128.4, 128.1, 127.5, 127.2, 124.1, 52.1, 46.0, 25.7. HRMS (ESI) calcd for C25H20ClO2 [M+H]+ 387.1152, found 387.1146.
(E)-2-(2-(4-Bromophenyl)-2-oxoethyl)-3-(chloro(phen-yl)methylene)-2-methyl-2,3-dihydro-1H-inden-1-one (3f, major): Yellow solid, 57.7 mg, 62% yield. m.p. 129~131 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.07~7.76 (m, 3H), 7.57~7.51 (m, 2H), 7.50~7.37 (m, 4H), 7.37~7.27 (m, 2H), 7.19 (d, J=4.4 Hz, 1H), 6.94 (d, J=7.6 Hz, 1H), 3.82 (d, J=18.4 Hz, 1H), 3.76 (d, J=18.4 Hz, 1H), 1.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.5, 196.4, 146.3, 144.6, 136.6, 135.8, 134.8, 134.1, 133.4, 131.9, 130.4, 128.7, 128.4, 128.2, 125.2, 124.2, 122.2, 121.2, 47.1, 45.4, 25.3. HRMS (ESI) calcd for C25H20BrClO2 [M+H]+ 465.0257, found 465.0268.
(E)-3-(Chloro(4-methoxyphenyl)methylene)-2-methyl-2-(2-oxo-2-phenylethyl)-2,3-dihydro-1H-inden-1-one (3g, major): Yellow solid, 30.8 mg, 37% yield. m.p. 110~112 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.83 (d, J=8.0 Hz, 1H), 7.99~7.81 (m, 2H), 7.78~7.73 (m, 1H), 7.65 (d, J=7.6 Hz, 2H), 7.57~7.51 (m, 2H), 7.45~7.34 (m, 3H), 7.34~7.27 (m, 1H), 3.77 (s, 3H), 3.37 (d, J=18.8 Hz, 1H), 2.85 (d, J=18.8 Hz, 1H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.2, 196.4, 159.9, 147.1, 135.8, 134.6, 133.2, 129.4, 128.4, 128.1, 127.5, 124.1, 113.9, 55.4, 52.2, 46.0, 25.7. HRMS (ESI) calcd for C26H22ClO3 [M+H]+ 417.1257, found 417.1258.
(E)-3-(Chloro(4-chlorophenyl)methylene)-2-methyl-2-(2-oxo-2-phenylethyl)-2,3-dihydro-1H-inden-1-one (3h, major): Yellow solid, 42.1 mg, 50% yield. m.p. 125~127 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.81 (d, J=8.0 Hz, 1H), 8.00~7.80 (m, 2H), 7.80~7.74 (m, 1H), 7.64 (d, J=7.6 Hz, 2H), 7.60~7.53 (m, 2H), 7.45 (d, J=7.2 Hz, 1H), 7.43~7.34 (m, 3H), 7.31 (d, J=8.0 Hz, 1H), 3.41 (d, J=18.8 Hz, 1H), 2.80 (d, J=18.4 Hz, 1H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 205.5, 196.2, 146.5, 139.2, 138.0, 135.8, 135.5, 135.1, 134.7, 133.5, 129.7, 128.6, 128.5, 128.2, 128.0, 127.5, 125.7, 124.2, 52.1, 46.0, 25.7. HRMS (ESI) calcd for C25H19Cl2O2 [M+H]+ 421.0762, found 421.0768.
(E)-3-((4-Bromophenyl)chloromethylene)-2-methyl-2-(2-oxo-2-phenylethyl)-2,3-dihydro-1H-inden-1-one (3i, major): Yellow solid, 55.8 mg, 60% yield. m.p. 124~126 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.92~7.83 (m, 2H), 7.76 (d, J=7.6 Hz, 1H), 7.61~7.49 (m, 2H), 7.47~7.42 (m, 1H), 7.40 (d, J=9.2 Hz, 1H), 7.35 (d, J=8.0 Hz, 2H), 7.33~7.28 (m, 3H), 7.22~7.16 (m, 1H), 7.07 (d, J=7.6 Hz, 1H), 3.74 (s, 1H), 3.30 (s, 1H), 1.35 (d, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 195.4, 143.6, 143.1, 137.1, 134.0, 131.9, 131.6, 129.6, 129.5, 128.7, 128.5, 128.4, 128.3, 125.2, 123.9, 121.2, 46.9, 45.2, 25.3. HRMS (ESI) calcd for C25H19BrClO2 [M+H]+ 465.0257, found 465.0263
(E)-3-(Chloro(phenyl)methylene)-2-methyl-2-(tosyl-methyl)-2,3-dihydro-1H-inden-1-one (3j): Yellow solid, 74.1 mg, 85% yield. m.p. 134~136 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.92 (d, J=4.0 Hz, 1H), 7.82~7.76 (m, 2H), 7.61 (s, 1H), 7.57~7.47 (m, 4H), 7.47~7.38 (m, 3H), 7.21 (d, J=8.0 Hz, 2H), 3.50 (d, J=16.0 Hz, 1H), 3.15 (d, J=12.0 Hz, 1H), 2.40 (s, 3H), 1.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 203.5, 147.1, 144.6, 139.1, 137.2, 135.5, 131.3, 129.9, 128.5, 127.9, 127.4, 124.2, 61.9, 51.2, 25.7, 21.6. HRMS (ESI) calcd for C25H22ClO3S [M+H]+ 437.0978, found 437.0986.
(E)-3-(Methoxy(phenyl)methylene)-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5a, major): Yellow solid, 41.7 mg, 65% yield. m.p. 105~107 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.37 (d, J=8.0 Hz, 1H), 8.22 (d, J=8.4 Hz, 1H), 7.81 (d, J=7.6 Hz, 1H), 7.75 (d, J=8.0 Hz, 1H), 7.63~7.67 (m, 1H), 7.48~7.50 (m, 2H), 7.37~7.38 (m, 2H), 3.43 (s, 3H), 2.85 (d, J=18.4 Hz, 1H), 2.06 (d, J=18.4 Hz, 1H), 1.81 (s, 3H), 1.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.0, 205.0, 152.3, 148.7, 134.9, 134.1, 134.0, 130.6, 130.0, 129.6, 128.6, 127.3, 127.1, 123.7, 121.40, 55.9, 50.6, 48.6, 29.1, 26.1. HRMS (ESI) calcd for C21H21O3 [M+H]+ 321.1491, found 321.1490.
(E)-3-(Methoxy(4-methoxyphenyl)methylene)-2-meth-yl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5b, major): Yellow oil, 29.4 mg, 42% yield. 1H NMR (400 MHz, CDCl3) δ: 8.36 (d, J=8.0 Hz, 1H), 7.80 (d, J=7.6 Hz, 1H), 7.61~7.65 (m, 1H), 7.33~7.37 (m, 1H), 7.21 (d, J=7.6 Hz, 2H), 6.99 (d, J=7.6 Hz, 2H), 3.89 (s, 3H), 3.41 (s, 3H), 2.86 (d, J=18.4 Hz, 1H), 2.15 (d, J=18.4 Hz, 1H), 1.83 (s, 3H), 1.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.2, 204.2, 159.6, 151.5, 148.0, 134.0, 133.1, 130.5, 129.8, 126.3, 126.2, 125.3, 122.8 120.5, 113.1, 54.9, 54.6, 49.9, 47.8, 28.3, 25.2. HRMS (ESI) calcd for C22H23O4 [M+H]+ 351.1596, found 351.1593.
(E)-3-(Methoxy(m-tolyl)methylene)-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5c, major): colorless oil, 45.5 mg, 68% yield. 1H NMR (400 MHz, CDCl3) δ: 8.36 (d, J=8.0 Hz, 1H), 7.80 (d, J=7.6 Hz, 1H), 7.73 (d, J=7.2 Hz, 1H), 7.61~7.66 (m, 1H), 7.35~7.39 (m, 2H), 7.22~7.20 (m, 2H), 3.35 (s, 3H), 3.20 (d, J=18.4 Hz, 1H), 2.83 (d, J=18.4 Hz, 1H), 2.53 (s, 3H), 2.17 (s, 3H), 1.48 (s, 3H). HRMS (ESI) calcd for C22H23O3 [M+H]+ 335.1647, found 335.1642.
(E)-3-(Methoxy(thiophen-2-yl)methylene)-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5d, major): Yellow solid, 22.8 mg, 35% yield. m.p. 98~100 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.22 (d, J=8.0 Hz, 1H), 7.75 (d, J=8.0 Hz, 2H), 7.52~7.59 (m, 2H), 7.21~7.23 (m, 1H), 7.15~7.18 (m, 1H), 3.46 (s, 3H), 3.21 (d, J=18.4 Hz, 1H), 2.35 (d, J=18.4 Hz, 1H), 2.04 (s, 3H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.7, 206.0, 147.7, 143.9, 134.9, 134.2, 130.7, 130.5, 129.0, 127.8, 127.2, 125.7, 123.9, 123.5, 55.8, 50.6, 50.5 29.8, 23.0; IR (KBr) ν: 1712.8, 1599.6, 1524.1, 1359.6 cm-1. HRMS (ESI) calcd for C19H19SO3 [M+H]+ 327.1055, found 327.1054.
(Z)-3-((4-(tert-Butyl)phenyl)(methoxy)methylene)-6-chloro-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5e, major): yellow oil, 45.2 mg, 55% yield. 1H NMR (400 MHz, CDCl3) δ: 7.67 (s, 1H), 7.49 (d, J=7.2 Hz, 2H), 7.26-7.23 (m, 2H), 7.07 (d, J=8.8 Hz, 1H), 6.12 (d, J=8.4 Hz, 1H), 3.63 (d, J=17.2 Hz, 1H), 3.36 (s, 3H), 3.18 (d, J=17.6 Hz, 1H), 2.05 (s, 1H), 1.39 (s, 9H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.0, 206.0, 153.3, 153.0, 146.7, 136.2, 133.9, 132.5, 130.5, 126.3, 124.6, 123.4, 123.4, 121.1, 56.1, 50.8, 50.6, 34.1, 31.4, 29.8, 22.9. HRMS (ESI) calcd for C25H28ClO3 [M+H]+ 411.1727, found 411.1724.
(E)-6-Chloro-3-((4-chlorophenyl)(methoxy)methylene)-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5f, major): Colorless solid, 48.2 mg, 62% yield. m.p. 108~110 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.29 (d, J=8.4 Hz, 1H), 7.75 (s, 1H), 7.58 (d, J=8.4 Hz, 1H), 7.27~7.29 (m, 2H), 7.17~7.22 (m, 2H), 3.41 (s, 3H), 2.88 (d, J=18.4 Hz, 1H), 2.07 (d, J=18.4 Hz, 1H), 1.84 (s, 3H), 1.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.3, 204.8, 151.4, 146.7, 135.3, 134.8, 133.4, 131.9, 131.8 130.3, 129.8, 129.7 128.3, 123.4, 121.1, 116.0, 115.8, 55.9, 50.8, 48.9, 29.1, 25.9. HRMS (ESI) calcd for C21H19Cl2O3 [M+H]+ 389.0711, found 389.0713.
(E)-6-Fluoro-3-(methoxy(phenyl)methylene)-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5g, major): Yellow oil, 38.6 mg, 57% yield. 1H NMR (400 MHz, CDCl3) δ: 8.33~8.37 (m, 1H), 7.48~7.50 (m, 3H), 7.42~7.41 (m, 2H), 7.34~7.37 (s, 2H), 3.41 (s, 3H), 2.83 (d, J=18.4 Hz, 1H), 2.46 (d, J=18.8 Hz, 1H), 2.06 (s, 3H), 1.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.2, 204.9, 162.1 (1JCF=247.6 Hz), 151.7, 144.8 (4JCF=2.4 Hz), 136.7 (3JCF=7.0 Hz), 136.8 133.8, 130.0, 129.9, 128.9, 128.6, 125.0 (3JCF=7.6 Hz), 122.2 (2JCF=23.0 Hz), 109.4 (2JCF=21.8 Hz), 55.9, 50.8, 49.2, 29.8, 26.0. 376; 19F NMR (376 MHz, CDCl3) δ: -114.0. HRMS (ESI) calcd for C21H20FO3 [M+H]+ 339.1396, found 339.1391.
(E)-3-(Methoxy(p-tolyl)methylene)-2-methyl-2-(2-oxo-propyl)-6-(trifluoromethyl)-2,3-dihydro-1H-inden-1-one (5h, major): Yellow oil, 41.8 mg, 52% yield. 1H NMR (400 MHz, CDCl3) δ: 7.96 (s, 1H), 7.36~7.26 (m, 5H), 3.42 (s, 3H), 2.86 (d, J=19.2 Hz, 1H), 2.46 (s, 3H), 2.13 (d, J=19.2 Hz, 1H), 2.06 (s, 3H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.0, 206.1, 154.7, 151.5, 140.1, 134.8, 131.1 (JCF=3.2 Hz), 130.3, 129.4, 128.5 (JCF=32.7 Hz), 127.5, 124.2 (JCF=270.8 Hz), 123.8, 121.0 (JCF=3.9 Hz), 120.8, 56.0, 50.8, 50.5, 29.7, 22.7, 21.6; 19F NMR (376 MHz, CDCl3) δ: -62.6. HRMS (ESI) calcd for C23H22F3O3 [M+H]+ 403.1521, found 403.1520.
(E)-1-(Methoxy(p-tolyl)methylene)-2-methyl-3-oxo-2-(2-oxopropyl)-2,3-dihydro-1H-indene-5-carbonitrile (5i, major): Yellow solid, 47.4 mg, 66% yield. m.p. 113~115 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.45 (d, J=8.4 Hz, 1H), 7.87 (s, 1H), 7.74 (d, J=8.0 Hz, 1H) 7.32~7.30 (m, 2H), 7.14 (d, J=7.6 Hz, 2H), 3.43 (s, 3H), 2.85 (d, J=18.8 Hz, 1H), 2.47 (s, 3H), 2.13 (d, J=19.2 Hz, 1H), 1.85 (s, 3H), 1.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 206.4, 206.0, 152.0, 140.6, 137.2, 135.0,130.6, 130.0, 129.5, 128.0, 127.8, 124.1, 120.8, 118.7, 110.0, 56.2, 50.9, 50.4, 29.7, 25.7, 21.7. HRMS (ESI) calcd for C23H22NO3 [M+H]+ 360.1600, found 360.1605.
(E)-5-Chloro-3-(methoxy(phenyl)methylene)-2-methyl-2-(2-oxopropyl)-2,3-dihydro-1H-inden-1-one (5j, major): yellow oil, 39.0 mg, 55% yield. 1H NMR (400 MHz, CDCl3) δ: 8.36 (s, 1H), 7.72 (d, J=8.0 Hz, 1H), 7.49~7.55 (m, 5H), 7.33 (d, J=8.0 Hz, 2H), 3.44 (s, 3H), 2.82 (d, J=18.8 Hz, 1H), 2.06 (d, J=19.2 Hz, 1H), 1.82 (s, 3H), 1.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.2, 204.2, 159.6, 151.5, 148.0, 134.0, 133.1, 130.5, 126.3, 126.2, 125.3, 122.8, 120.5, 113.1, 54.8, 54.6, 49.9, 47.8, 28.3, 25.2. HRMS (ESI) calcd for C21H20ClO3 [M+H]+ 355.1101, found 355.1104.
(E)-3-(Methoxy(phenyl)methylene)-2-methyl-2-(2-oxo-2-phenylethyl)-2,3-dihydro-1H-inden-1-one (5k, major): yellow oil, 45.9 mg, 60% yield. 1H NMR (400 MHz, CDCl3) δ: 8.43 (d, J=8.0 Hz, 1H), 7.87 (d, J=7.2 Hz, 1H), 7.62~7.65 (m, 2H), 7.40~7.53 (m, 6H), 7.22~7.26 (m, 4H), 3.39 (s, 3H), 3.29 (d, J=18.4 Hz, 1H), 2.76 (d, J=18.8 Hz, 1H), 1.20 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.0, 196.5, 152.3, 148.7, 136.1, 134.8, 134.1, 133.8, 133.0, 130.1, 129.4, 128.5, 128.4, 128.1, 127.1, 123.9, 123.7, 55.8, 48.6, 46.3, 23.1. HRMS (ESI) calcd for C26H23O3 [M+H]+ 383.1647, found 383.1643.
Supporting Information 1H NMR and 13C NMR spectra of products 3a~3j and 5a~5k. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(Lu, Y.)