


碳碳键的形成是有机合成中最重要的基本反应, 开发原子经济性高且对环境友好的绿色可持续发展方法来促成新的碳碳键生成, 一直以来都是有机化学界的热点研究课题[1]. 醇类化合物是化学工业中和实验室最常见的原料之一, 自然界里许多天然产物及生物原料(如氨基酸、甘油、糖、萜类的含氧衍生物等)都是含醇羟基的化合物, 拥有种类多样的分子结构. 利用醇脱氧产生烷基自由基在有机化学中极具价值. 然而由于醇的碳氧键键能较大(≈401.3 kJ/mol)[2], 解离能力弱, 不易活化, 氧原子配位能力很强, 在反应中不易以OH的形式离去, 更容易使O—H键断裂等, 很大程度上限制了醇的广泛应用. 对此, 科学家们往往采用预活化的方式将醇羟基转化为相对稳定的活性中间体, 比如草酸酯、碳酸酯、溴代物和缩醛等, 然后再断裂C—O键实现其官能团化反应.
光或者电与过渡金属相结合的合成策略在过去的十年中得到了巨大的发展和应用[3]. 金属光/电氧化还原催化结合了过渡金属催化强大的键形成能力和光/电诱导电子转移过程, 规避了传统合成中有机金属试剂的制备和使用, 具有反应装置简单、化学选择性高和官能团兼容性良好等优点, 一定程度上补充了传统的催化平台. 这一新颖的催化模式为有机反应提供新的反应途径, 利用温和的反应条件, 一些传统的具有挑战性sp2- sp3偶联反应已可以利用廉价金属催化剂来实现[4], 这为醇衍生物的C—O键活化芳基化提供了新的发展机会.
有不少综述对醇的脱羟基化反应进行报道. 2015年, 王从洋团队[5]报道了铼催化醇脱羟基衍生化反应; 2020年, Samec团队[6]报道了过渡金属催化下π活化醇的Suzuki-Miyaura型交叉偶联反应; 2021年, 张万斌团队[7]报道了镍催化醇衍生物构筑碳-碳键的偶联反应; 2022年, Lundberg团队[8]报道了醇及其衍生物C—O键活化电合成反应; 2023年, 吴劼团队[9]报道了草酸酯类化合物的自由基脱羟基化反应; 2024年, Srimani团队[10]报道了可见光光氧化还原催化醇、羧酸等原料C—O键直接活化生成烷基和酰基的转化反应. 关于经过自由基途径的光/电催化下的醇与芳基卤代物或缺电子芳香腈C(sp2)—C(sp3)的偶联反应并没有被系统性阐述报道. 本文将总结光/电促进的醇衍生物C—O键活化与芳基卤代物或缺电子芳香腈的偶联反应的研究进展, 按照酯类化合物、氮杂环卡宾加合物、膦氧加合物、溴代物以及缩醛等分类阐述, 并讨论了部分反应的机理.
1 酯类化合物作为醇的衍生物实现C—O键活化
1.1 草酸酯类化合物C—O键活化
在过去的半个世纪里, 过渡金属催化的碳-碳键偶联反应对合成化学产生了深远影响, 尤其是钯催化的交叉偶联方法. 这些方法使得将简单起始原料转化为复杂结构分子变得更为容易和高效. Heck、Negishi和Suzuki三位化学家因在这一领域的开创性工作获得了2010年诺贝尔化学奖[11]. 虽然钯催化的交叉偶联在sp2杂化的碳中心成键方面取得了巨大成功, 但在涉及sp3杂化的碳中心偶联时仍存在一些挑战和局限性. 这些局限性包括底物的选择范围较窄、反应条件要求较苛刻以及产率不稳定等.
镍催化在这一充满挑战的领域取得了实质性进展[12]. 与钯相比, 镍具有较小的原子半径和较低的电负性, 而且低价的镍物种具有更负的氧化还原电位, 通常使得镍催化剂在氧化加成或插入反应中具有更高的反应性. 它能激活更惰性的亲电体, 如烷基溴代物, 并且使用脂肪族配体可以抑制β-H消除的副反应的影响. 另一个关键点是, 与Pd(I)和Pd(III)相比, Ni(I)和Ni(III)的电子构型更为稳定. 镍催化剂经常涉及许多可相互转换的Ni(n+)可变氧化态的活性物质[包括Ni(0)、Ni(I)、Ni(II)和Ni(III)]. 相比之下, 涉及Pd(I)或Pd(III)活性物质的催化反应仍然发展较少, 可能需要特殊配体的促进或者与光催化或电催化结合[13]. 催化试剂的发展推动了有机合成方法的多样性, 但更为重要的是, 如何使用更为廉价丰富的起始原料成为化学家们研究的重点. 在这种情况下, 光氧化还原催化在旧键断裂后引入新的官能团过程中发挥着不可或缺的作用.
光氧化还原催化与过渡金属催化相结合, 称为金属光氧化还原催化. 在金属介导的键形成过程中, 可以选用简单的原料分子实现光催化底物活化, 在一定程度上补充了传统的催化平台. 2016年, MacMillan课题组[14]报道了利用草酰氯将醇一步转化为草酸酯作为自由基前体, 在可见光与金属镍协同催化下实现其与芳基溴代物C(sp2)—C(sp3)键的交叉偶联(Scheme 1). 该反应使用廉价金属镍作为催化剂, 可以实现一级醇2、二级醇3及环状醇4的脱氧芳基化反应. 该方法可以成功应用于天然甾体化合物5的官能团化, 并且作者使用4-羟基-N-叔丁氧羰基哌啶(6)为底物高效地合成了抗结核药物Q203的先导物7.
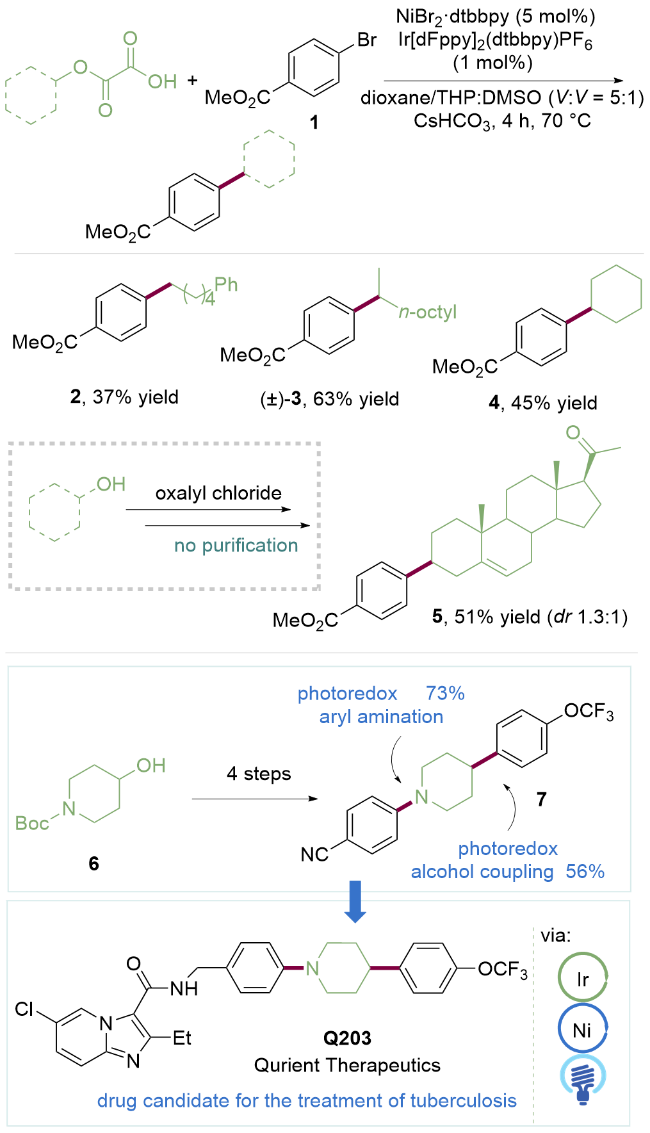
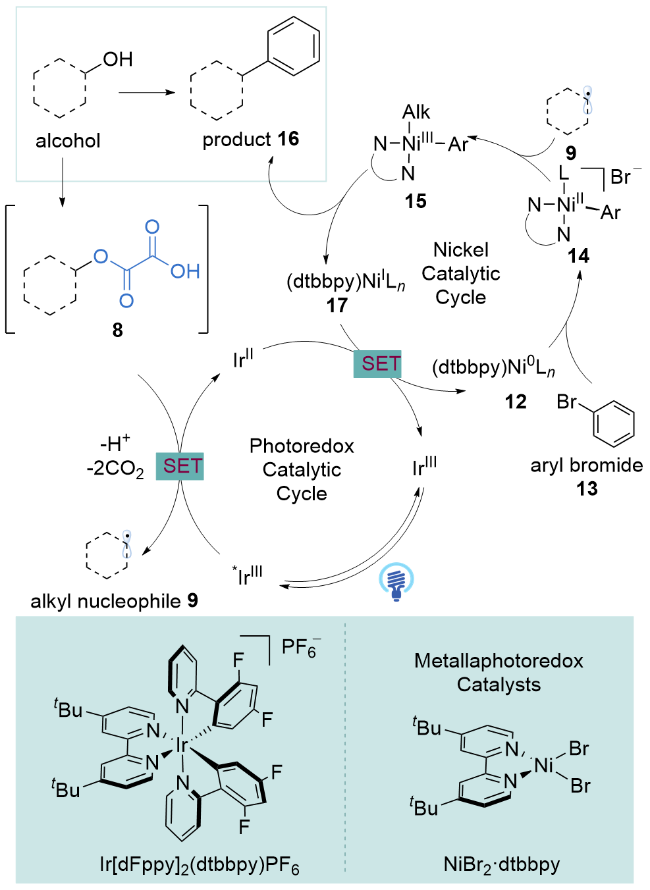
作者提出的反应机理如Scheme 2所示, 首先醇与草酰氯原位生成草酸酯8, 去质子化后的8氧化半波电位Ep/2=+1.26 V (vs SCE in CH3CN). 激发态的*IrIII催化剂($E_{\mathrm{red}}^{1 / 2}$=[*IrIII/IrII]=+1.1 V, vs SCE in CH3CN)可以将该草酸酯氧化, 脱去两分子二氧化碳后生成热力学更稳定的烷基自由基9. 同时, Ni0配合物12与芳基溴代物13氧化加成生成芳基NiII配合物14. 烷基自由基与该中间体发生自由基加成反应得到(烷基)(芳基)NiIII中间体15, 进一步还原消除得到NiI配合物17和芳基化产物16. 随后NiI配合物与还原性的IrII催化剂发生单电子转移反应得到IrIII催化剂和Ni0配合物12, 完成两个催化循环. 该反应利用草酸酯作为醇的衍生物产生烷基自由基, 实现了可见光促进的双亲电试剂C(sp2)—C(sp3)交叉偶联反应, 开辟了醇作为C(sp3)部分参与金属光氧化还原策略的先河, 大大拓展了C(sp3)部分的底物范围.

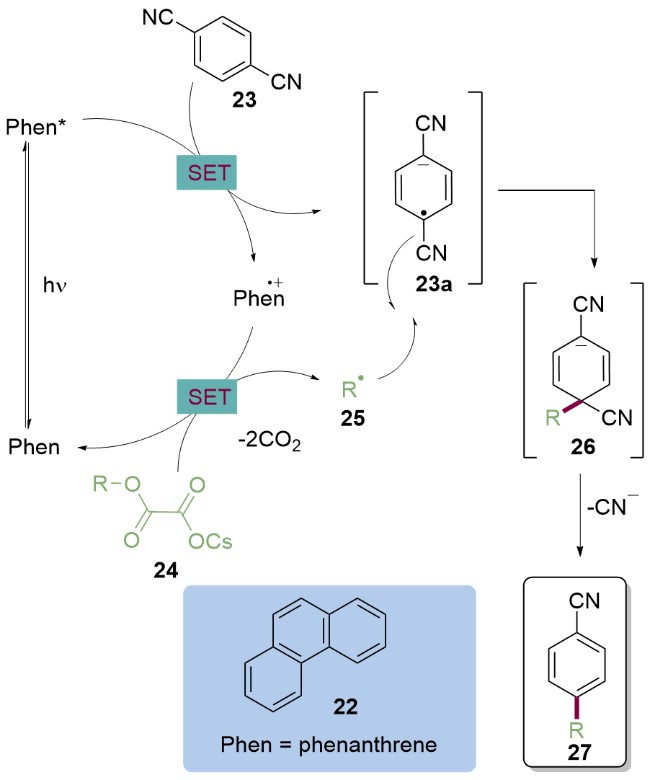
作者提出的反应机理如Scheme 4所示, 首先光催化剂菲22在紫外光的照射下生成激发态菲( =[*Phen/Phen•+]=-2.1 V, vs SCE), 然后与对苯二腈23 ($E_{\mathrm{red}}^{1 / 2}$=[DCB/DCB•−]=-1.6 V, vs SCE)发生单电子转移, 将其还原为对苯二腈自由基阴离子23a和菲自由基阳离子. 后者作为强氧化剂($E_{\mathrm{red}}^{1 / 2}$=[Phen•+/Phen]=1.5 V, vs SCE)与草酸铯24发生单电子转移, 脱去两分子二氧化碳生成烷基自由基25, 然后快速与对苯二腈自由基阴离子23a发生自由基偶联, 随后氰基离去得到C(sp2)—C(sp3)产物27.
对于多取代烯烃常常存在构型异构体. Z∶E构型的烯烃的物化性质和生物活性均表现出较大差异, 所以烯烃制备的Z∶E选择性控制在有机合成方法学中具有很重要的研究意义. 现已报道的烯烃的Z∶E选择性制备方法主要集中在Wittig反应、取代烷烃消除反应、烯烃异构化反应、烯烃复分解反应、烯烃偶联反应、炔烃加成反应和卡宾偶联反应等. 然而, 大多文献报道的高选择性制备热力学稳定的E-烯烃的方法比较多, 而高选择性制备Z-烯烃的方法相对比较欠缺. 而且对于烯烃的Z∶E选择性研究存在很多的困难, 通过催化剂或者载体控制实现烯烃的顺反选择性依然存在一定的挑战. 因此, 如何突破热力学控制, 实现高选择性制备Z-烯烃具有广泛的研究和应用意义[20].
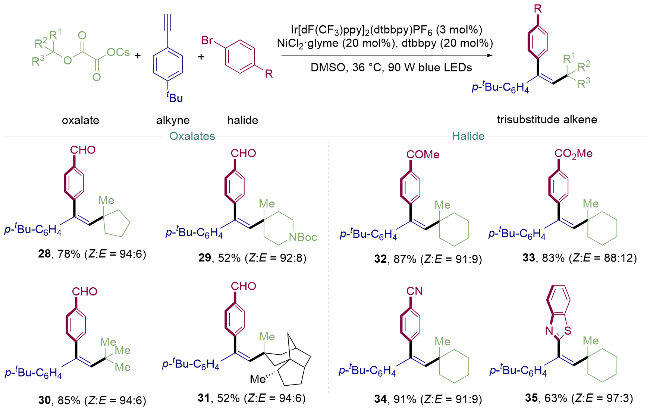
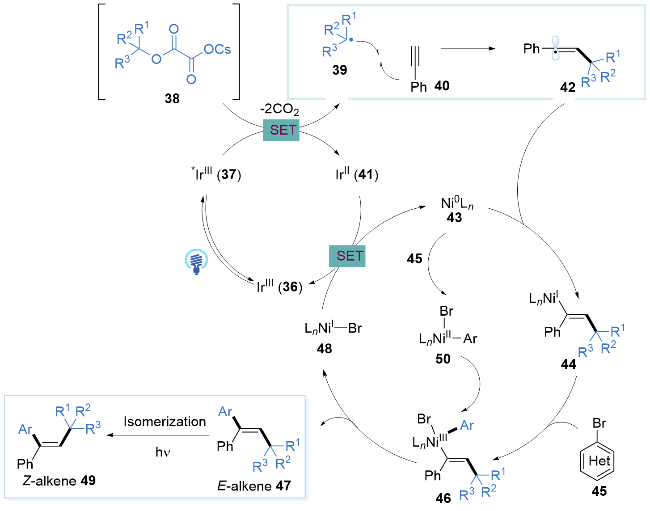
2018年, 储玲玲课题组[21]结合金属光协同催化与光致烯烃异构化策略[22], 使用三级烷基醇草酸酯、2-苯炔和芳基溴代物, 通过光氧化还原催化三组分偶联, 一锅法实现了末端炔烃的同面双官能化团反应. 这种催化模式以优异的合成效率和较高的立体选择性将末端炔烃转化为高度官能团化的三取代Z-烯烃. 该温和的条件能够兼容大量官能团, 具有广泛的底物范围, 包括末端炔烃、芳基溴代物和草酸烷基酯. 底物烷基醇可以使用普通环状醇28、杂环醇29、叔丁醇30以及具有多环结构的(+)-雪松醇31, 吸电子基团取代(如乙酰基32、酯基33及氰基34)的溴苯均可兼容, 芳杂环溴代物如2-溴苯并噻唑35也展现出良好的产率以及优秀的异构选择性(Scheme 5).
在该金属光氧化还原催化三组分偶联反应中(Scheme 6), 可见光激发IrIII (36)产生激发态*IrIII (37), 通过单电子转移还原烷基草酸酯38, 脱去两分子二氧化碳的同时产生烷基自由基39, 后者与炔烃40进行选择性自由基加成得到共振式更加稳定的烯基自由基42. 对于镍催化的循环可以通过两种反应路径得到目标产物49. 一种路径是烯基自由基42先与Ni0配合物43结合得到NiI配合物44, NiI配合物44再与芳基卤化物45发生氧化加成. 另一种路径是芳基溴代物45先与Ni0配合物氧化加成得到Ar-NiII-Br配合物50, 然后NiII物种再与烯基自由基42反应. 两种路径都会得到共同的NiIII反应中间体46, 随后发生还原消除得到交叉偶联的芳基烷基化产物47以及NiI物种48. 后者通过单电子转移氧化IrII (41)完成催化循环. 最后Z-烯烃47在光照下发生烯烃异构化得到反热力学的产物Z-烯烃49. 该催化模式通过连续的碳-碳键形成步骤, 为末端炔烃高选择性地转化成为三取代Z-烯烃提供了一个高效、温和的催化平台.
烯烃类化合物由于其便宜易得的特点, 在有机合成的发展中占有极为重要的地位[24]. 碳碳双键作为普遍的前手性基团, 可通过一步法转化为手性基团, 且可以合成高度复杂的化合物, 因此烯烃的双官能团化反应非常重要.
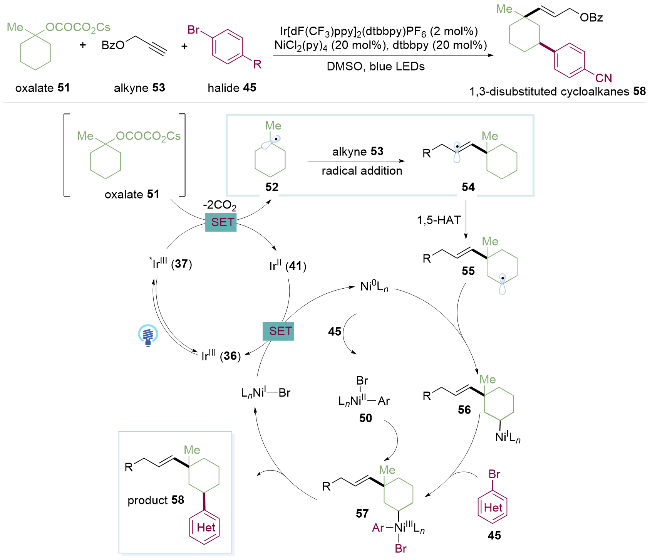
烯烃的双官能团化反应作为一类重要的有机化学反应, 不仅可以经济、有效地一步合成多位点反应产物, 而且可以将起始原料转化为多种含有生物活性或药物活性的化合物, 同时还为构建化学结构的多样性提供了更多的方法. 因此, 发展烯烃的双官能团化反应一直是有机化学中的研究热点[25]. 烯烃的双官能团化三组分反应是有机合成中通过一步构建两个连续的碳碳键, 由简单分子合成复杂分子的直接策略. 其中过渡金属催化的烯烃与碳-亲核试剂的共轭交叉偶联是最高效、最强大的合成方法之一[26]. 但是, 这种策略在很大程度上受到双组分反应和β-H消除等副反应的影响, 特别是当使用未活化的烯烃作为反应底物时. 基于自由基机理的方法为解决这些问题提供了一个巧妙的代替手段. 具体来说, 通过金属催化剂与光催化剂的协同催化作用, 包括自由基与烯烃的加成, 镍催化剂辅助偶联实现选择性分子间的双官能团化反应.
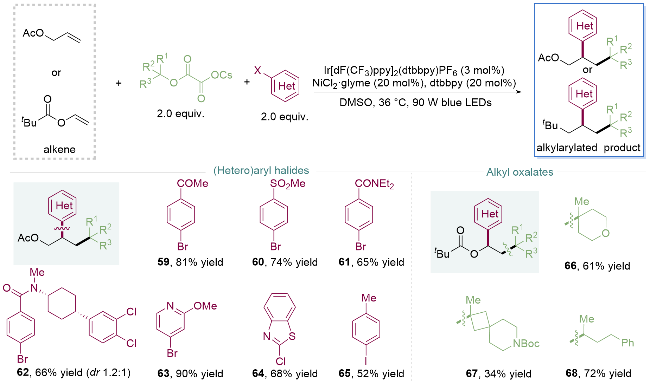
2019年, 储玲玲课题组[27]使用与醇、炔烃和溴苯的三组分偶联反应(Scheme 5)同样的反应条件, 一步构建两个连续的碳-碳键, 实现了烯烃的同面选择性烷基芳基化. 值得注意的是, 未活化的烯烃, 吸电子基团取代的芳基溴代物如羰基59、砜60或酰胺61, 杂环如对溴吡啶63、苯并噻唑氯代物64, 给电子基团取代的碘苯65, 以及单环叔醇、螺环叔醇和链状叔醇组成的烷基草酸酯, 在该金属光氧化还原的温和条件下都能顺利进行反应, 突破了传统过渡金属催化下使用亲核和亲电试剂与烯烃的共轭交叉偶联. 另外作者利用该反应实现了对舍曲林衍生物62的官能化, 展现出优秀的官能团兼容性和底物适用范围(Scheme 8).
1.2 甲磺酸酯类化合物C—O键活化
烷基磺酸酯, 特别是一级烷基醇的磺酸酯, 容易与溴离子等进行原位卤素交换, 形成高活性的烷基溴物种来参与偶联反应. 基于卤素交换, 许多镍催化烷基卤化物参与的交叉偶联反应也能推广到烷基磺酸酯底物.
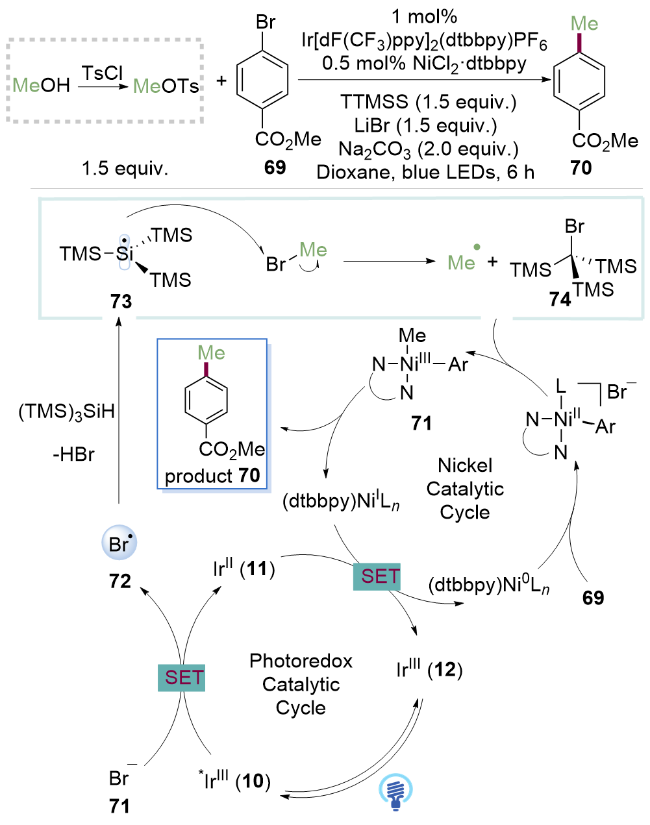
2016年, MacMillan课题组[28]报道了可见光与金属镍协同催化烷基溴与芳基溴化合物的亲电交叉偶联反应, 实现了一例对甲基苯磺酸甲酯的脱氧芳基化. 该反应使用[Ir[dF(CF3)ppy]2(dtbbpy)]PF6和NiCl2•dtbbpy作为催化剂, 溴化锂(LiBr)作为溴源与对甲苯基磺酸甲酯反应, 原位生成一溴甲烷(CH3Br). 硅自由基通过溴自由基与三(三甲基硅基)硅烷(TTMSS)发生卤原子转移(XAT)生成. 在硅自由基介导的可见光氧化还原条件下, 生成高活性的甲基自由基, 继而与芳基镍物种发生C(sp2)—C(sp3)偶联反应.
作者所使用的磺酸酯试剂相比传统的偶联反应所使用的有机硼酸试剂、格氏试剂及有机锌试剂等更加易于制备, 且在水氧条件下稳定. 在该光氧化还原循环中(Scheme 9), 芳基NiII物种有可以解离的配体溴负离子(71), 其还原电势为 =+0.80 V (vs SCE in DME). 激发态的*IrIII催化剂( =[*IrIII/IrII]=+1.21 V, vs SCE in CH3CN)可以将溴负离子氧化, 生成溴自由基72. 后者与三(三甲基硅基)硅烷(TTMSS)反应生成更稳定的硅基自由基73. 接下来, 由于Si—Br键的键解离能(BDE=401 kJ/mol, Me3Si—Br)相对于C—Br键的键解离能(289 kJ/mol, 溴乙烷)更高, 硅自由基可以攫取一溴甲烷的溴原子, 得到亲核性的甲基自由基. 同时, Ni0配合物与芳基溴代物69氧化加成生成芳基NiII配合物. 甲基自由基与该中间体发生自由基加成反应得到(甲基)(芳基)NiIII中间体, 进一步还原消除得到NiI配合物和产物70. 随后NiI配合物与还原性的IrII催化剂发生单电子转移反应得到IrIII催化剂和Ni0配合物, 完成两个催化循环. 该反应利用对甲苯磺酸甲酯作为甲醇的衍生物实现C—O键活化芳基化反应, 为脂肪醇C—O活化方法的进一步研究提供了新思路.
1.3 苯甲酸酯类化合物C—O键活化
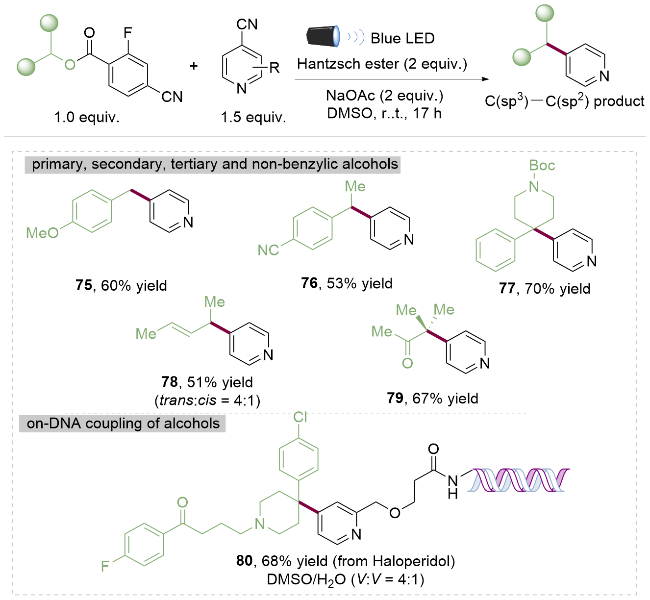
苯甲酸酯类化合物相对较为稳定, 并且容易制备, 可使用苯甲酸与醇类化合物在酸催化条件下一步缩合而成. 如果能实现其高效的转化反应, 则苯甲酸酯类化合物可以在合成策略中发挥更为关键的作用. 2022年, Nappi课题组[29]报道了光催化下使用吸电子基团取代的苯甲酸酯作为醇的衍生物, 实现其与对氰基吡啶的C(sp2)—C(sp3)偶联反应. 作者通过外加汉斯酯作为光氧化还原催化的电子给体, 在光照下实现了对一级醇75、二级醇76与三级醇77的吡啶化反应, 而且烯丙醇78和α-羰基醇79也能进行反应, 具有广泛的底物适用性和官能团耐受性(Scheme 10). 不仅如此, 在水作为共溶剂的条件下能够实现药物分子氟哌啶醇(haloperidol, 80)的DNA片段修饰. 显示出该方法在药物合成中巨大的应用潜力.
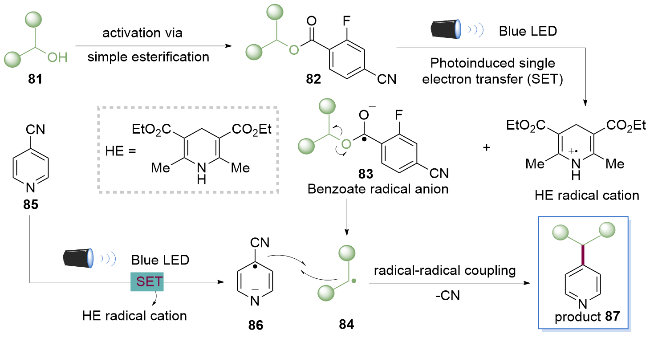
作者使用缺电子的苯甲酸酯82与汉斯酯(HE)形成一对电子给体-受体复合物(EDA complex). 在可见光的照射下, 这一对复合物发生分子间的电子转移, 得到苯甲酸酯自由基阴离子83, 随后发生β-断裂得到烷基自由基84和副产物羧酸盐. 同时对氰基吡啶85被HE还原为自由基阴离子86, 再与烷基自由基84发生自由基偶联反应, 离去氰基得到产物87, 该反应进一步拓展了醇的活化方式(Scheme 11).
1.4 二氢吡啶羧酸酯类化合物C—O键活化
芳基糖基化合物相比于O-和N-糖苷类似物具有更好的体内稳定性. 因此, C-芳基糖苷作为候选药物被广泛研究, 并应用于化学生物学领域. 然而上述氧化还原活性酯类化合物包括草酸酯、甲磺酸酯及苯甲酸酯等并未应用于糖基化反应, 可能是由于相应的糖基酯不稳定, 易于发生β-消除等副反应.
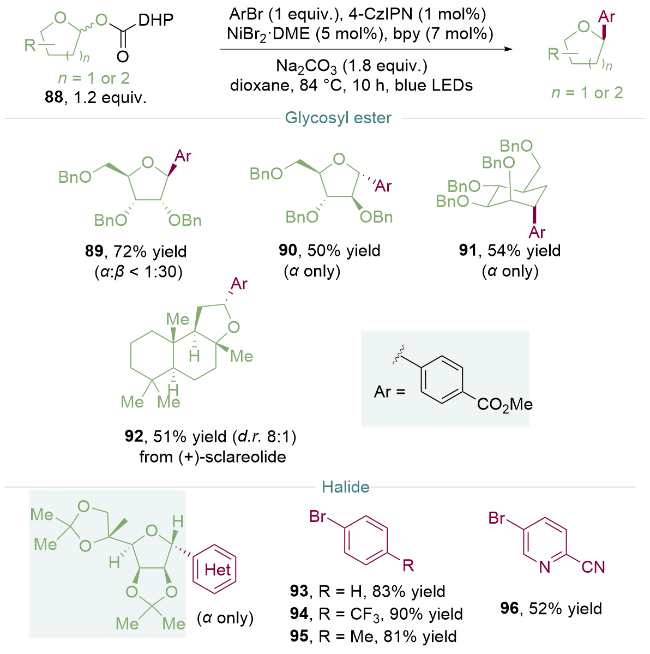
2021年, 刁天宁团队[30]开发了一种由二氢吡啶(DHP)羧酸糖基酯88介导的糖类化合物C—O键活化方法. 该酯类化合物在光氧化还原催化条件下可以稳定地生成氮自由基, 与随后的烷氧基羰基自由基脱羧结合, 产生烷基自由基, 实现糖类化合物的芳基化反应. 该体系对于呋喃糖(89, 90)、吡喃糖91和衍生天然产物分子[92, (+)-sclareolide]均可适用, 并且对于苄氧基保护的D-呋喃核糖, 反应主要经过β-芳基化的途径生成产物89; 而底物为D-阿拉伯呋喃糖时C-芳基化反应则发生了立体选择性的改变, 只生成α-异构体90, 反映了C(2)取代基对立体化学结果的主导作用. 此外, 芳环上带有不同取代基的芳基溴化物(93~95)以及含杂环吡啶的溴化物96均可顺利反应, 展现出广泛的底物适用范围. 同时糖基酯的易制备和稳定性显示出该方法在药物化学中巨大的应用潜力(Scheme 12).
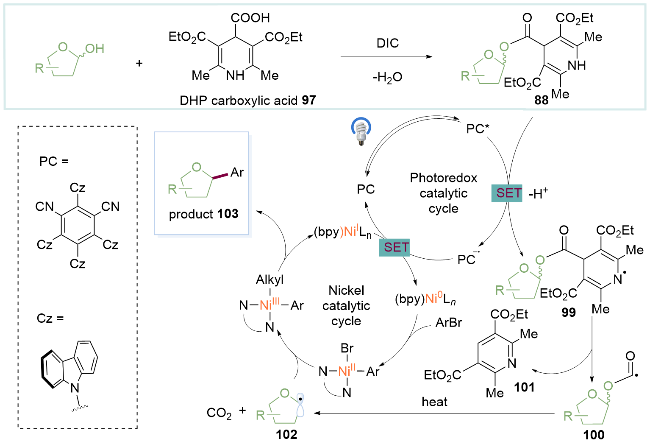
作者预先将醇与二氢吡啶羧酸97在二异丙基碳二亚胺(DIC)的条件下缩合得到具有氧化还原活性的DHP糖基酯88. 后者在去质子化后被激发态的光催化剂氧化, 得到DHP糖基酯氮自由基99. 随后Hantzsch吡啶101离去得到烷氧基羰基自由基100, 在加热的促进下脱去一分子二氧化碳, 生成的糖基自由基102进入镍催化循环中完成碳碳键的构建. 这一机制为醇的CO键活化提供了一种新的策略(Scheme 13).
1.5 黄原酸酯类化合物C—O键活化
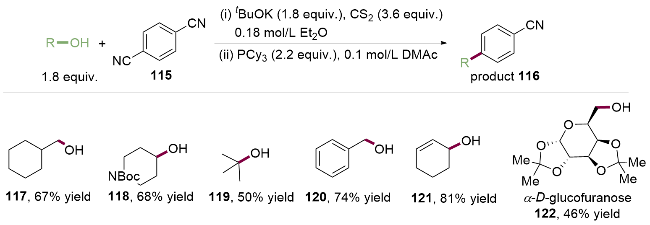
将醇转化为硫代酯类中间体, 然后断裂C—O键得到醇的脱羟基产物的反应, 被称为Barton-McCombie脱羟基反应[31]. 2017年, Molander课题组[32]通过将苄醇预先活化为黄原酸苄酯104, 实现了镍/光催化黄原酸苄酯与溴代芳烃的偶联反应(Scheme 14). 与传统利用BF3/ O2引发体系不同, 作者使用市售可得的仲丁基三氟硼酸钾盐111作为醇活化的自由基前体引发剂. 该反应体系对于苯环上带有不同取代基的苄醇和酚类化合物均可适用(105~107), 但C(sp2)部分仅限于吸电子取代的芳基溴代物(108~110). 在光作用下, 仲丁基三氟硼酸钾盐111产生的仲丁基自由基112优先活化黄原酸苄酯中的碳氧键, 产生苄基自由基113, 然后进入镍催化循环中, 完成碳碳键的构建. 作者巧妙地控制两种不同自由基的相对速率, 为醇类衍生物作为烷基自由基前体进行C(sp2)—C(sp3)交叉偶联反应, 增添了新的维度.
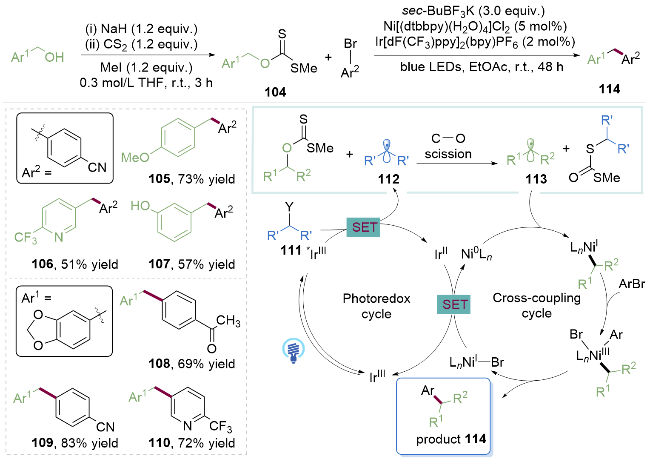
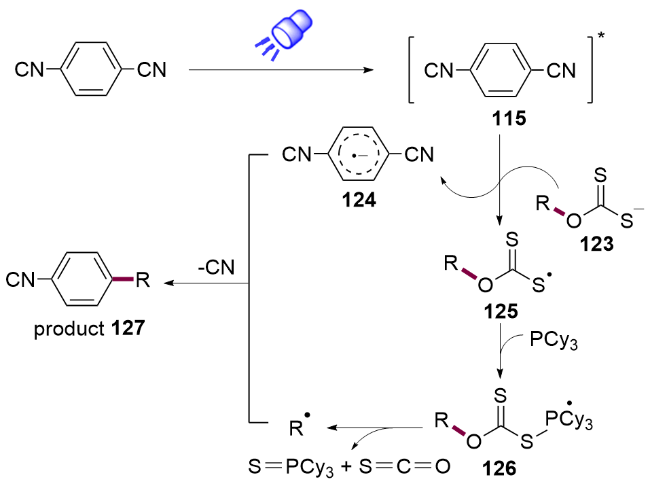
作者提出的机理如Scheme 16所示. 首先在光照下产生激发态的对苯二腈115 ( =+2.55 V, vs SCE), 通过单电子转移氧化黄原酸钾盐123得到对苯二腈自由基阴离子124和硫自由基125. 后者被三环己基膦捕获产生磷自由基126, 随后发生β-断裂, 得到的烷基自由基与对苯二腈自由基阴离子124发生自由基脱氰偶联反应, 构建C(sp2)—C(sp3)键.
2 氮杂环卡宾(NHC)介导的烷基醇C—O键活化
醇直接脱氧交叉偶联反应的最大挑战在于C—O键裂解前醇的原位活化, 这是因为该步骤需要较高的热力学驱动力. 2021年, MacMillan课题组[34]基于N-杂环卡宾与极性O—H键之间发生可逆缩合与α-氨基C—H键均裂等方面的研究[35], 利用N-杂环卡宾盐对游离醇进行原位活化, 通过金属光氧化还原催化的策略, 成功地实现了游离烷基醇与芳基卤化物的脱氧芳基化反应(Scheme 17). 该反应不仅具有广泛的底物适用性, 而且能实现抗糖尿病药物捷诺维(129, Januvia)的合成, 以及抗癌药物分子紫杉醇类似物(130, Taxol variant)和降血脂药辛伐他汀类似物(131, Simvastatin variant)的脱氧芳基化反应, 更加突出了这种脱氧转化对实现复杂底物官能团化反应的强大功能.
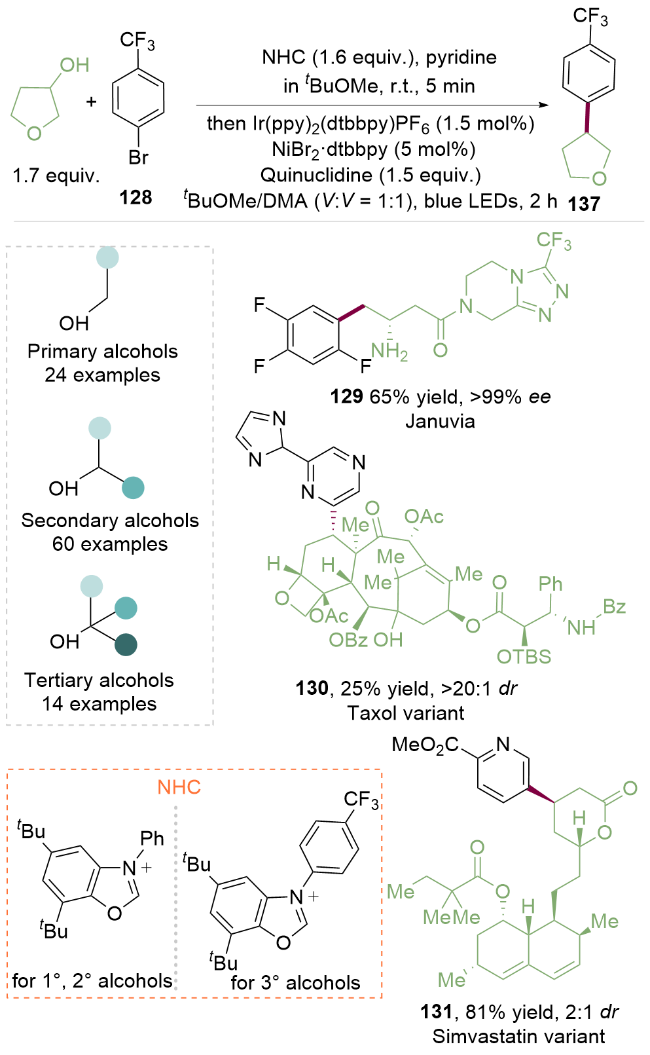
作者考察了一系列氮杂环卡宾盐的发现, 极度缺电子的N-芳基苯并噁唑盐能够抑制与醇形成加合物后解离的倾向, 在光催化剂氧化后得到N阳离子自由基中间体133 (pKa≈10)[36], 在碱的作用下去质子化后得到α-氨基自由基134. 由于邻位的3个杂原子(N、O、O), 其发生β-断裂在动力学上势能很低; 同时在该基元步骤上发生C—O键均裂后生成的副产物氨基甲酸酯136具有强大的热力学驱动力, 并产生烷基自由基135. 随后, 烷基自由基135被NiII物种捕获生成NiIII中间体, 再发生还原消除得到NiI配合物和C(sp2)—C(sp3)偶联产物137. 随后NiI配合物与还原性的IrII催化剂发生单电子转移反应得到IrIII催化剂和Ni0配合物, 完成两个催化循环. 该方法为醇活化为C(sp3)自由基提供了强大的方法, 有望在化学合成及药物合成中进一步广泛应用(Scheme 18).
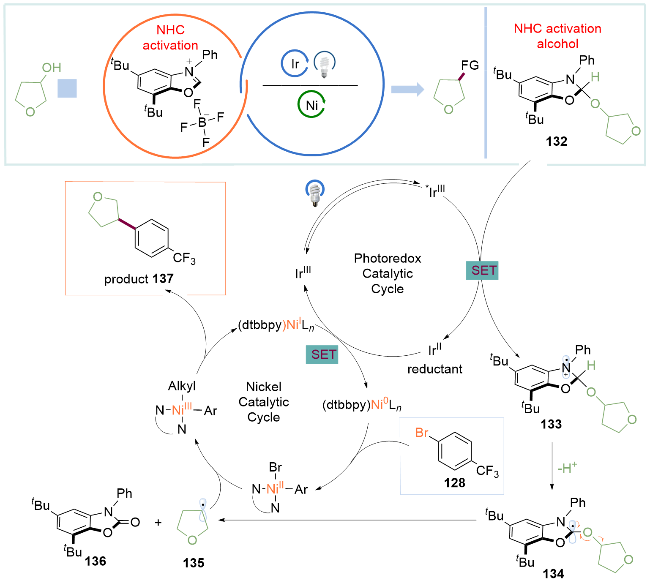
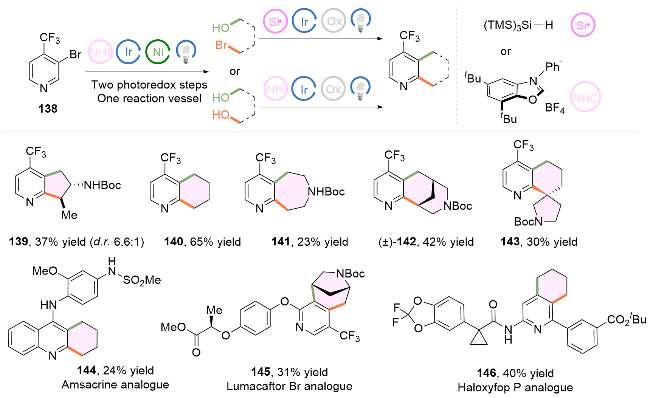
半饱和杂芳烃通常具有较高比例的C(sp3)成分, 表现出靶标结合亲和力和特异性. 然而, 半饱和环骨架的合成通常需要多步合成. 2024年, MacMillan课题组[37]借助上述氮杂环卡宾介导的烷基醇C—O键活化的方法, 发展了一种简单的、模块化的双自由基偶联-关环策略来高效构建半饱和杂环体系(Scheme 19). 自由基前体底物可选用溴醇或者二醇, 具有广泛的底物适用范围. 该转化将金属光氧化还原C(sp2)—C(sp3)交叉偶联与分子内Minisci型自由基环化相结合, 快速构建了传统方法难以合成的分子骨架, 如五元环139、六元环140、七元环141、双环142以及螺环143. 作者还实现对复杂药物分子如抗癌药物安吖啶类似物(144, Amsacrine variant)、治疗囊性纤维化的芦马卡托类似物(145, Lumacaftor variant)以及除草剂高效氟吡甲禾灵类似物(146, Haloxyfop P variant)的合成, 验证了该方法优秀的兼容性.
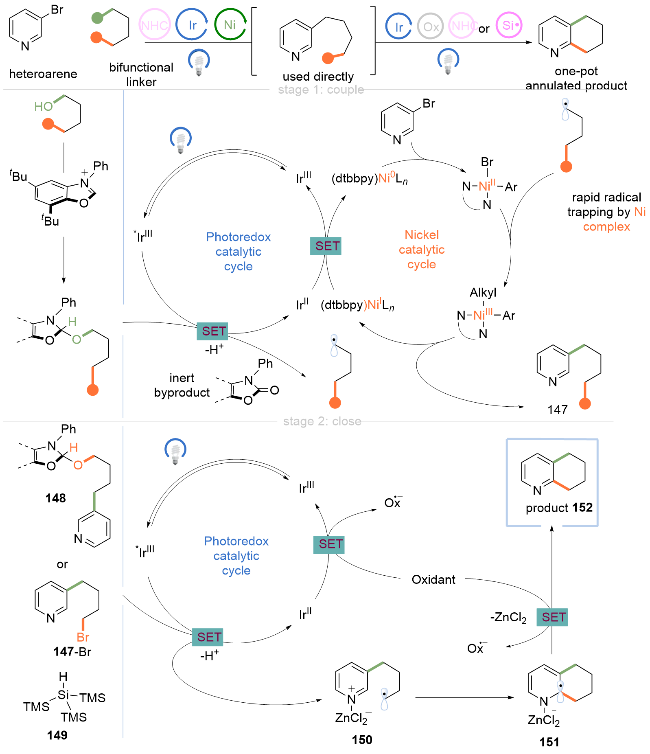
作者提出该偶联-关环反应机理如Scheme 20所示. (1)金属光氧化还原偶联: 与上述机理(Scheme 18)类似, 首先醇通过C(sp2)—C(sp3)偶联得到脱氧芳基化产物147. (2)光氧化还原Minisci环化: 当使用二醇作为反应试剂时, 中间产物147被额外的NHC/吡啶活化, 形成活性中间体148. 在光照下, 激发态的Ir*催化剂氧化148, 释放出烷基自由基150. 当使用溴醇作为反应试剂时, 需要将TTMSS 149加入到反应混合物中, 烷基自由基150通过溴醇与TTMSS 149之间发生卤原子转移(XAT)生成. 通过使用氯化锌作为Lewis酸, 促进分子内Minisci环化反应生成中间体151, 最后被单电子氧化得到产物152. 此外, IrII物种可以通过单电子氧化得到IrⅢ, 完成光催化循环. 该策略由试剂控制自由基形成过程, 具有高区域选择性和立体特异性, 可用于药物骨架的后期官能团化, 避免了冗长的从头合成半饱和环的过程.
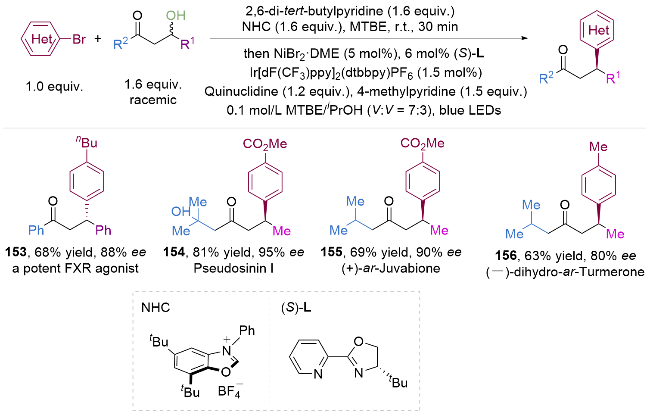
3 膦试剂助力烷基醇C—O键活化
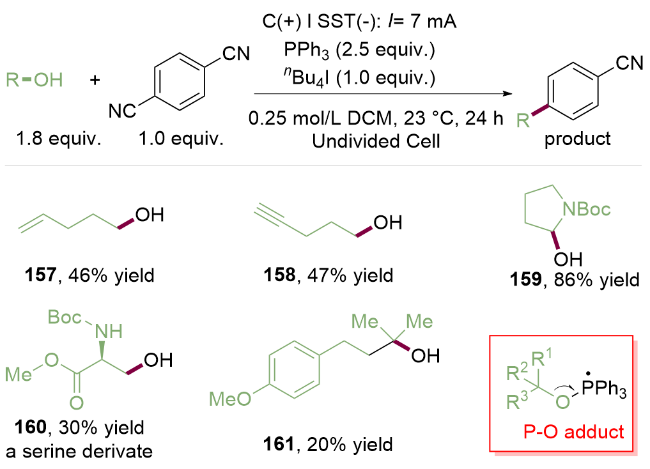
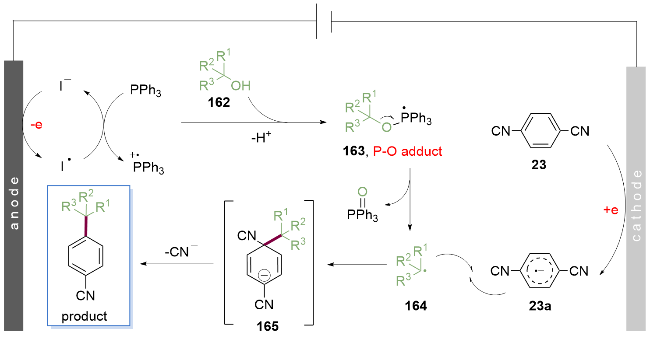
作者提出了如Scheme 23所示机理, 首先碘负离子在阳极经历单电子氧化生成碘自由基, 其作为有效的中间介质迅速将三苯基膦氧化, 后者与游离醇162反应去质子化后得到烷氧基三苯基膦自由基163, 由于强的P=O键键能作为驱动力, 该自由基163发生β-断裂得到烷基自由基164. 同时, 在阴级一侧, 对苯二腈23发生阴极还原, 得到的芳香自由基阳阴离子23a与烷基自由基164发生自由基偶联反应, 产生中间体165, 最后氰基离去, 得到目标产物.
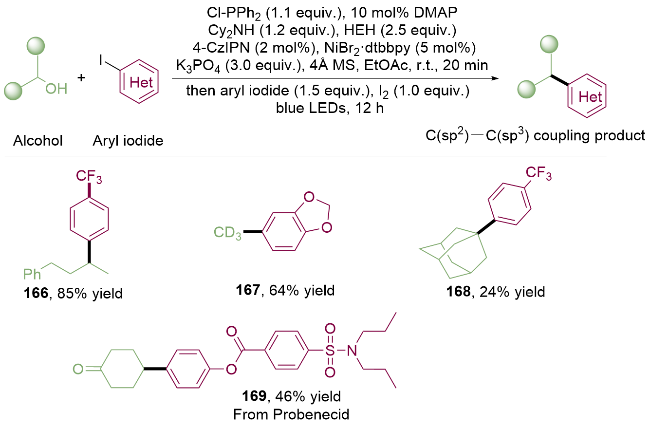
2024年, 徐涛课题组[40]利用廉价易得的二苯基氯化膦作为原位活化试剂, 在有机碱二环己基胺(Cy2NH)的作用下, 与醇形成关键中间产物P—O缩合物. 以碘单质作为外源自由基来源, 得到的膦自由基中间体发生β-断裂产生烷基自由基, 以芳基碘代物作为C(sp2)来源, 通过金属光氧化还原策略高效地实现了C(sp2)—C(sp3)键的构筑. 该方法可以方便地实现一级醇、二级醇、三级醇的芳基化(166~168). 不仅如此, 作者以商业化的氘代甲醇作为烷基自由基前体, 可以实现芳基碘代物167的氘代甲基化. 该反应体系可以兼容丰富的官能团, 同时能够实现对复杂药物丙磺舒的后期修饰(169, from Probenecid), 在药物化学上具有一定的应用价值(Scheme 24).
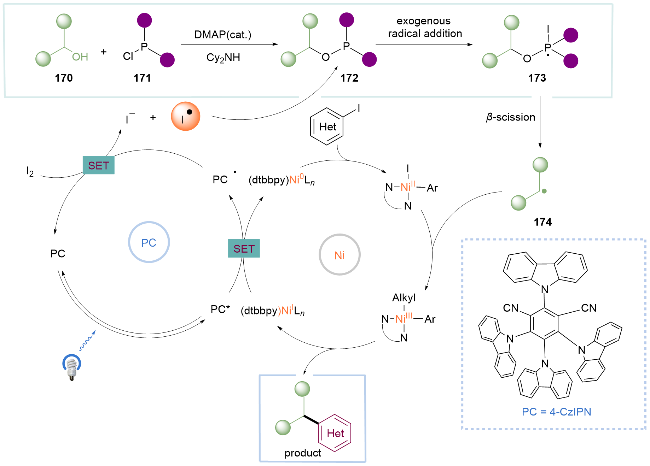
作者提出的机理如Scheme 25所示, 首先醇170与二苯基氯化膦171在4-二甲氨基吡啶(DMAP)/Cy2NH的作用下缩合得到P-O加合物172. 由光催化剂还原产生的外源碘自由基加成至P-O加合物172上产生膦自由基173, 随后发生β-断裂生成烷基自由基174. 174被NiII物种捕获, 进一步发生还原消除得到C(sp2)—C(sp3)产物. 该反应发展了膦试剂助力活化烷基醇的脱氧芳基化反应的方法.
4 烷基醇原位溴化作为醇的衍生物实现C—O键活化
电化学合成法通过非常规的机制途径发现新的化学转化, 其作为一种绿色、高效、可控的合成方法, 在有机合成等领域具有重要的应用前景. 2021年, 李超团队[41]通过电化学方法将阳极的Appel反应[42]和镍催化阴极交叉亲电偶联反应相结合, 体系中三苯基膦与游离的溴离子将醇原位活化为关键的中间产物烷基溴代物, 其被单电子还原生成烷基自由基进入镍催化循环作为C(sp3)部分与芳基溴代物发生C(sp2)—C(sp3)偶联反应. 其关键在于匹配氧化还原电解对反应固有的氧化还原电势, 以及协调阳极氧化、阴极还原和金属催化循环的反应速率. 该反应可以实现伯醇和仲醇的直接脱氧转化, 并且具有良好的官能团兼容性和较高的产率, 对含酰亚胺的底物醇175、含羰基的底物醇176、含胺的底物醇177、含氰基的底物醇178、 茚醇179和含酯基的底物醇180均可适用. 该方法还能实现药物分子和天然产物分子雌激素受体调节剂奥培米芬(181, ospemifene)及叶绿醇(182, phytol)的脱氧官能团化, 为复杂药物分子和天然产物分子的后期修饰和改造提供了良好的方法. 此外, 芳环上带有不同取代基的芳基溴化物(183~186)以及杂环体系如吡啶187、吲哚188、苯并呋喃189和苯并噁唑190等均可顺利反应, 展现出广泛的底物适用范围, 具有广泛的应用前景(Scheme 26).

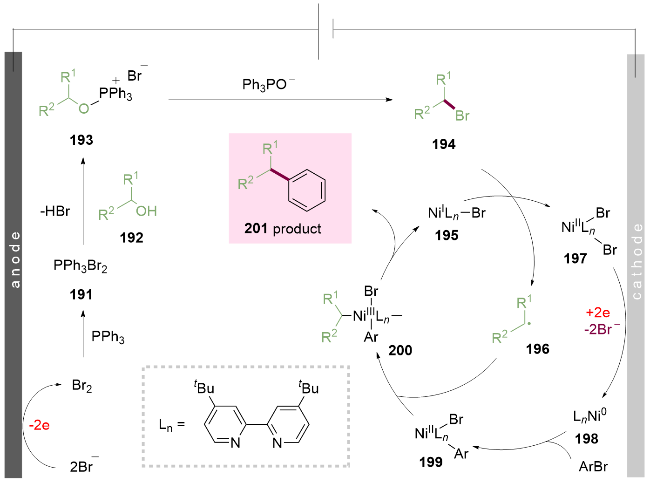
作者提出的机理如Scheme 27所示, 溶液中的溴负离子首先在电解池的阳极被氧化失去两个电子生成溴单质, 溴单质与三苯基膦反应生成二溴三苯基膦191. 紧接着191与底物醇192反应生成Mitsunobu中间体193, 被溴离子进攻的同时脱去一分子三苯氧膦得到烷基溴194. 后者被一价镍195还原生成烷基自由基196. 同时, 二价的镍盐197在阴极得到两个电子被还原为零价镍198. 这一还原过程保持了电解池正负极的氧化还原平衡和电解质的电荷平衡. 还原得到的零价镍198紧接着氧化插入到芳基溴之间形成二价的镍物种199, 此二价镍物种再与烷基自由基196结合生成三价的镍物种200, 最终还原消除得到目标偶联产物201. 阳极氧化释放的溴负离子可以通过传质和静电作用回到阳极, 再次与醇偶联得到烷氧基三苯基溴化鏻193, 这使得相比一般的Appel反应, 该反应仅需催化量的溴离子. 该方法为从游离醇中获取烷基自由基提供了新的活化方法, 有望在电化学合成中得到广泛应用.
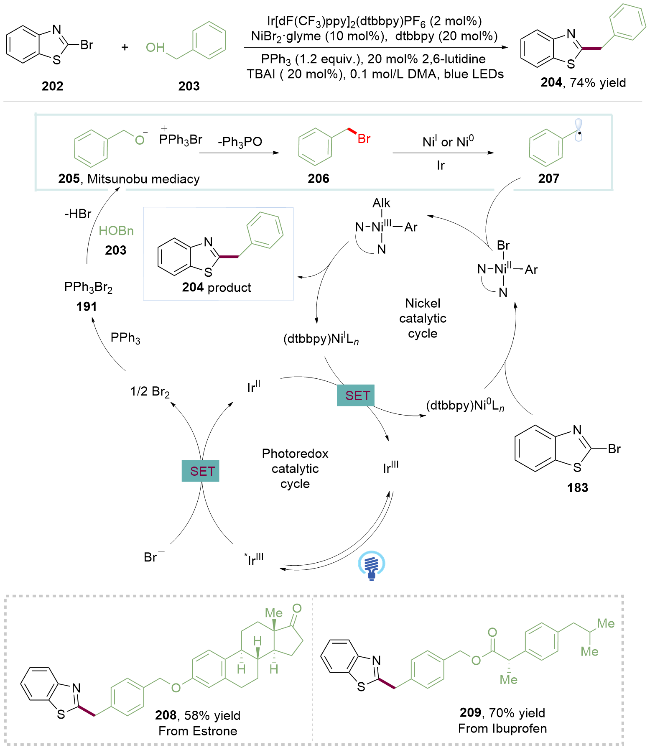
2024年, 王惠飞课题组[43]同样采用PPh3将醇原位溴化的方式, 在金属光催化下实现了苄醇的脱氧芳基化. 在光照下, 激发态的Ir*催化剂与溴负离子发生单电子氧化生成溴单质, 其迅速活化三苯基膦产生二溴三苯基膦191, 随后二溴三苯基膦191与苄醇203缩合产生苄氧基三苯基鏻盐中间体205, 后者被溴离子进攻的同时脱去一分子三苯氧膦得到苄溴206. 后者可以被攫溴或者被催化剂单电子还原得到苄基自由基207进入镍催化循环构筑C(sp2)—C(sp3)键. 该反应条件温和, 能够应用于复杂药物和生物活性分子的后期官能团化, 如雌酚酮类似物(208, Estrone analogue)和布洛芬类似物(209, Ibuprofen analogue)的脱氧官能团化, 具有良好的官能团耐受性, 有望为药物分子合成提供一个新的平台(Scheme 28).
5 缩醛作为醇的衍生物实现C—O键活化
甲基化反应是有机合成和药物化学领域进行结构修饰的一类重要反应. 在药物分子结构中引入甲基有助于提高其与生物大分子结合亲和力、生物溶解度和代谢稳定性, 这种现象被称为“神奇的甲基效应”[44].
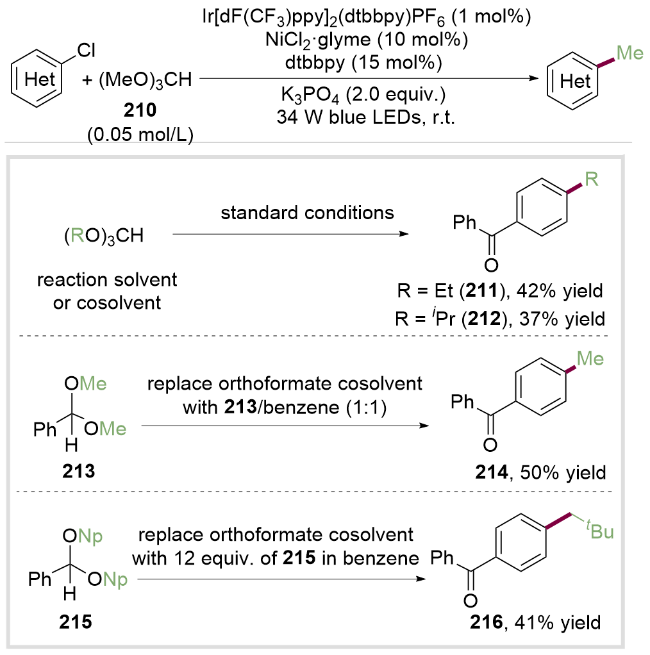
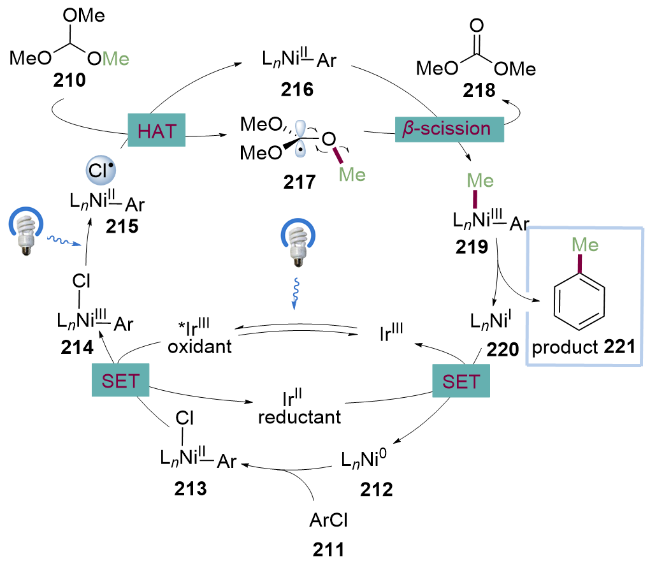
作者提出的反应机理如Scheme 30所示. 首先芳基氯化物211与Ni0配合物212氧化加成生成芳基NiII配合物213. 该络合物被激发态的*IrIII催化剂氧化为芳基NiIII络合物214, 214在光照下发生LMCT过程[46]生成氯自由基, 氯自由基进一步与原甲酸三甲酯210发生氢原子转移过程(HAT)生成原甲酸三甲酯叔碳自由基217, 随后发生β-断裂得到甲基自由基和碳酸二甲酯218. 甲基自由基随后被NiII配合物216捕获, 得到NiIII配合物219之后, 发生还原消除得到C(sp2)—C(sp3)偶联产物221及NiI配合物220. 得到的NiI配合物220可以将二价铱配合物氧化成三价铱配合物, 同时NiI配合物220被还原为Ni0配合物212, 完成催化循环. 该方法为从醇的衍生物获取甲基自由基以及其他脂肪族自由基提供了新的方法, 有望在化学合成及药物合成中得到广泛应用.
2022年, Doyle课题组[47]受到上述工作的启发, 利用烷基醇与苯甲醛缩合制得的缩醛作为烷基自由基前体, 实现了缩醛与芳基溴代物或芳基氯代物的C(sp2)—C(sp3)偶联反应. 反应机理包括氢原子转移过程(HAT)、光催化和镍催化的三者循环, 其提升产率的关键在于匹配三者之间的循环速率. 作者利用数据科学挑选各种代表性的芳基溴代物底物来评价该金属光策略偶联反应. 在该温和条件下磺酰胺(222, 223)、氰基(224、225)、酯基(226)和氟(227)等官能团均可兼容, 而且对于二取代和多取代的芳基溴代物也能获得中等至优秀的产率, 展现了缩醛介导的烷基醇C—O键活化应用于C(sp2)—C(sp3)偶联反应的强大功能(Scheme 31).
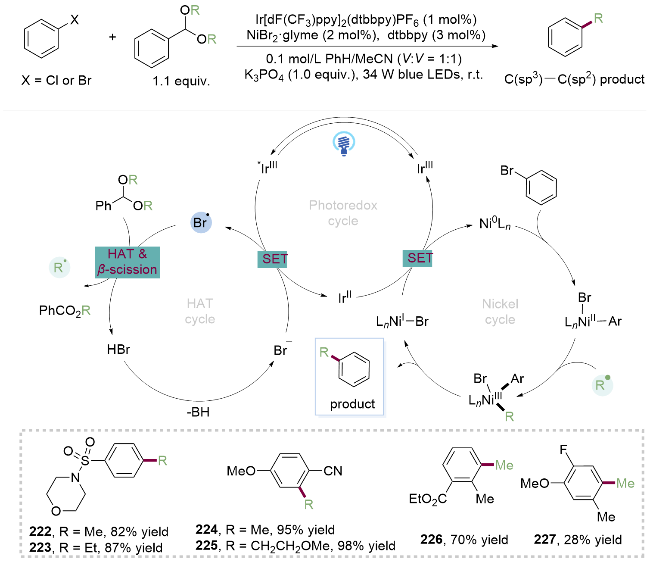
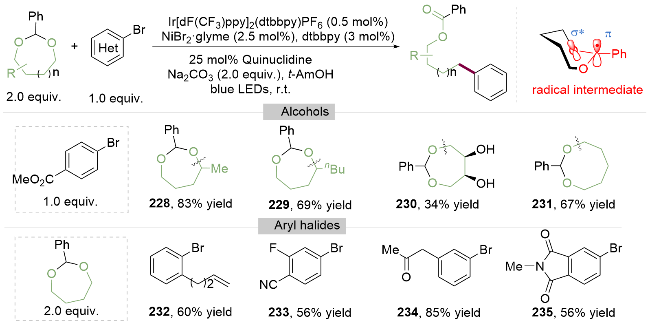
2022年, Martin课题组[48]报道了环状缩醛的选择性C—O键断裂芳基化反应. 作者结合密度泛函理论指出其关键在于大环(n>6)缩醛的构象灵活性和较小的环张力, 只有当环状缩醛在某一特定旋转角度的构象下, 其叔碳自由基的p轨道与相邻氧间连的C(sp3)—O反键轨道发生重叠产生σ*-p超共轭效应(Scheme 32), 形成相对势能更低的过渡态, 才能引发环状缩醛的C(sp3)—O键断裂. 该金属光氧化还原催化方法具有广泛的环状缩醛和芳基溴代物的适用范围. 其中, 对于非对称环状缩醛(228, 229), 由于仲碳自由基稳定性高于伯碳自由基, 其断键位置在带有更多取代基的C(sp3)—O键位点. 而以直接与氧相连的碳上无取代基的环状缩醛作为底物,时, 同样能够实现C(sp3)—O键活化, 并且缩醛底物中含有游离的醇羟基, 并不会受到影响(230~231). 在该光/镍双催化模式下, 含双键(232)、氰基和氟原子(233)、羰基(234)及酰亚胺235等官能团的取代的芳基溴代物均可反应, 为实现脂肪醇的C—O键活化提供了新的思路.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 总结与展望
综上所述, 由于镍原子半径较小, 具有多种可变的价态, 可以巧妙地与光/电结合为双催化循环模式. 多种基于自由基机理的金属光/电催化策略已经成为构筑碳-碳键的重要手段. 天然存在且稳定易得的醇相较于其他原料(卤代物、羧酸等)作为反应底物, 更加经济和绿色环保, 因此近年来受到了化学家的广泛关注和研究. 这一策略充分体现了绿色环保的理念, 并在复杂药物或天然分子的后期修饰当中展示了巨大的应用前景. 但由于醇的碳氧键键能较大, 化学家们通常采用预活化的方式, 将醇预活化为相对稳定并且具有氧化还原活性的中间体, 如酯类化合物、氮杂环卡宾加合物、膦氧加合物、溴代物以及缩醛等醇类衍生物. 可以预见, 未来会开发出更多潜在的醇活化试剂, 为实现醇作为烷基自由基前体参与镍催化C(sp2)—C(sp3)偶联反应提供一种简便、经济的新方法, 同时为醇类化合物转化为附加值更高的化学产品提供一条更加经济和高效的途径.
(Zhao, C.)