


1 Introduction
Organic azides are valuable intermediates in organic synthesis and as a result various methods have been developed for their synthesis.[1] Traditionally, α-azidoketones are prepared by direct nucleophilic substitution of α-halo ketone with azide ions.[2] Alternative methods are based on the reaction of an alkene or enol ether with an oxidizing agent in the presence of an azide source (Scheme 1).[3] However, most of these methods still require a large excess of expensive and hazardous oxidants, which would increase cost and environmental concerns, and limit their practical utility. From a green and sustainable points of view, the direct utilization of O2 as both oxidant and oxygen source represents an ideal methodology for the construction of oxygen-containing compounds. In 2018, Yang and co-workers[4] reported a facile visible-light enabled method for the construction of α-azidoketones via metal- free Rose Bengal and (PhSe)2 co-catalyzed oxyazidation of styrene with molecular oxygen. In 2021, Zhang and co- workers[5] reported an electrochemical synthetic route to access α-azido ketones from styrene with molecular oxygen. Although these two methods are relatively mild, the reaction substrates primarily focus on styrene derivatives and do not extend to alkyl-substituted olefins. Therefore, the development of a simple, efficient, and environmentally friendly method for the construction of various α-azido- ketones is still highly desirable.
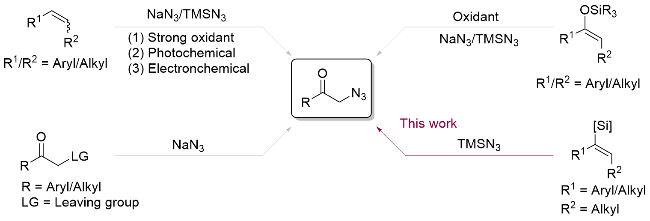
Alkenyl silanes are highly attractive intermediates due to their versatility in organic synthesis and materials science.[6] Since the presence of 3d empty orbitals of silicon atoms has the α-effect on stable α-C radical, alkenyl silanes can be regarded as effective radical acceptor.[7] In 1993, Isoe and co-workers[8] reported an electron-promoted aerobic oxythiolation of alkenyl silanes with PhSH to deliver the α-phenylthiol ketone. In 2003, Oshima and co-workers[9] reported that an Et3B-promoted radical addition followed aerobic oxidation of alkenyl silanes with alkyl iodides to afford ketones. Although the oxygenation of alkenyl silanes to obtain the corresponding α-phenylthio carbonyl compounds and ketones has been reported, using alkenyl silanes as substrates to construct C—N bond has not been reported. Based on our previous efforts in the construction of the C—N bond and aerobic oxidation reactions,[10] herein, we report a visible light-promoted aerobic oxidation of alkenyl silanes with TMSN3 to afford α-azido- ketones.
2 Results and discussion
At the beginning of our study, hex-1-en-2-yldiphenyl- silane (1a) was used as a model substrate, TMSN3 as a nitrogen source, Acr+$\mathrm{MesClO}_{4}^{-}$ as a photocatalyst and MeCN as a solvent under the irradiation of 8 W blue LEDs for 18 h in the presence of an O2 balloon. Initially, the reaction was conducted, delivering the final product in 71% yield (Table 1, Entry 1). However, screening a series of photocatalysts, the reactions afforded 3a in 13%~53% yields (Entries 2~5). After the optimization of the solvents, no better results were obtained (Entries 6~8). The yield was slightly reduced to 63% when the loading of Acr+$\mathrm{MesClO}_{4}^{-}$ was decreased to 2 mol% (Entry 9). Although the yield of 3a was increased to 80% with 10 mol% Acr+$\mathrm{MesClO}_{4}^{-}$, 5 mol% Acr+$\mathrm{MesClO}_{4}^{-}$ was chosen to further extend the application of this reaction (Entry 10).
Table 1 Variation of reaction conditions a |
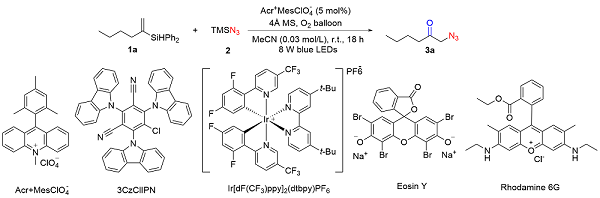
| Entry | Variation from the standard conditions | Yieldb/% |
|---|---|---|
| 1 | None | 71 (63) |
| 2 | 3CzClIPN as photocatalyst | 53 |
| 3 | Ir[dF(CF3)ppy]2(dtbpy)PF6 as photocatalyst | 32 |
| 4 | Eosin Y as photocatalyst | 13 |
| 5 | Rhodamine 6G as photocatalyst | 36 |
| 6 | DCM as solvent | 17 |
| 7 | THF as solvent | 0 |
| 8 | DMF as solvent | 0 |
| 9 | 2 mol% Acr+$\mathrm{MesClO}_{4}^{-}$ | 63 |
| 10 | 10 mol% Acr+$\mathrm{MesClO}_{4}^{-}$ | 80 |
a Reaction conditions: 1a (0.3 mmol), 2 (2.0 equiv.), Acr+$\mathrm{MesClO}_{4}^{-}$ (5 mol%) in MeCN (0.03 mol/L) and irradiation with 8 W blue LEDs under the atmosphere of oxygen at room temperature for 18 h. b Yields were determined by 1H NMR with TMSPh as an internal standard, and isolated yield was in the parenthesis. |
Under the optimized conditions, the substrate scope was explored as shown in Table 2. The linear aliphatic vinyl-silanes could be reacted to afford 3a~3e in 50%~72% yields. (1-Cyclohexylvinyl)diphenylsilane could be converted to the desired products 3f in 54% yield. It is worth noting that internal vinylsilanes could react smoothly to deliver the products 3g~3h in 51%~54% yields. The diphenyl(1-phenylvinyl)silane with a methyl group on para-, ortho-positions could be tolerated giving the corresponding products 3i~3j in 38%~42% yields. The methoxy group at para-position on aryl rings displayed moderate reactivity to afford the targeted products 3k in 54% yield. Moreover, the reactions of disubstituted styryl silanes and heterocyclic silanes bearing 3-chloro-4-methyl- phenyl, 3-thienyl and benzo[b]thiophen-5-yl could react with TMSN3 smoothly to produce the products 3l~3n in 37%~46% yields. The substrate containing bioactive molecule such as niflumic acid 3o could be also successfully accommodated, further demonstrating the utility of the approach. Furthermore, the various silane groups, such as dimethylphenyl silyl and diphenylmethyl silyl were found to be suitable for this transformation.
Table 2 Substrate scopea |
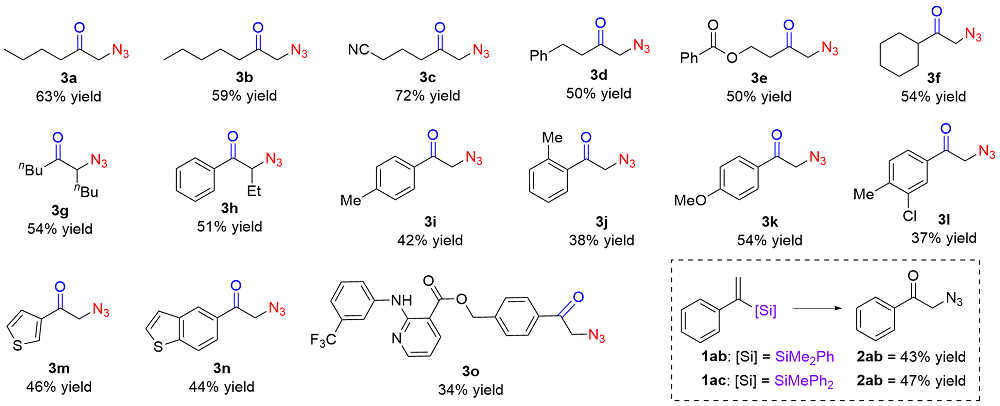 |
a Reaction conditions: alkenyl silanes (0.3 mmol), TMSN3 (2.0 equiv.), Acr+$\mathrm{MesClO}_{4}^{-}$ (5 mol%) and 4 Å MS (w=100% based on 1) in MeCN (0.03 mol/L) and irradiation with 8 W blue LEDs under the atmosphere of oxygen at room temperature for 18 h. Isolated yields were given. |
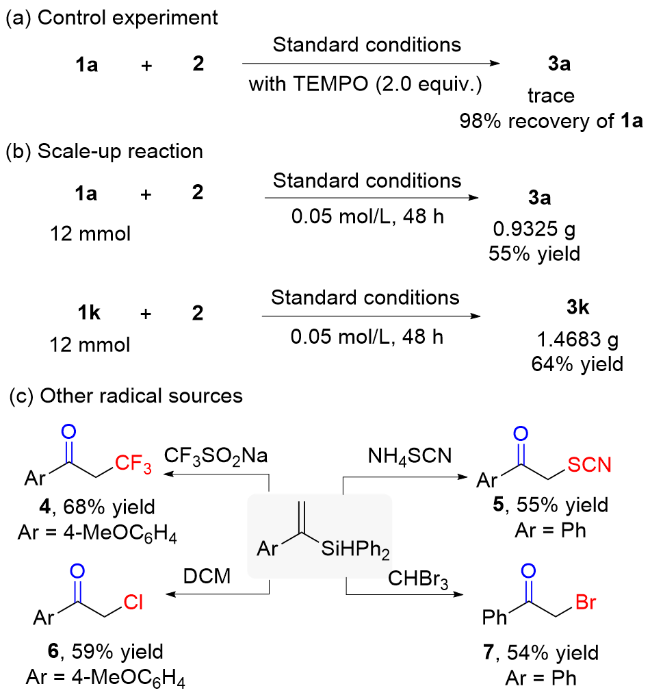
The reaction in the presence of 2,2,6,6-tetramethyl-piperidinooxy (TEMPO) afforded a trace amount of 3a with 98% recovery of 1a (Scheme 2a), which indicated that the reaction likely proceeds through a radical process. In addition, to further explore the scalability of this protocol, the gram-scale reactions were conducted by reacting 1a and 1k with 2 to afford targeted compounds in 55% and 64% yields (Scheme 2b), respectively. To explore the diversity of the reaction types, various radical sources, including trifluoromethyl radical, thiocyanate radical, bro- mide radical, chlorine radical, participated well in the reaction, affording the carbonyl compounds 4~7 in 54%~68% yields (Scheme 2c).
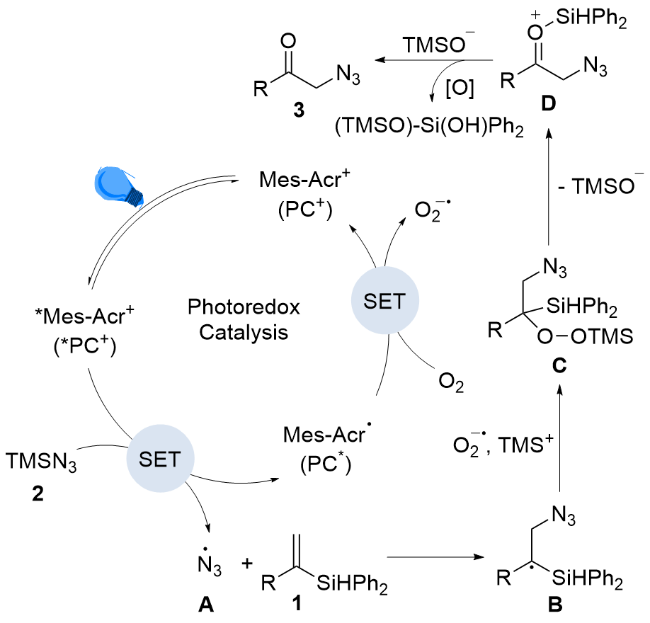
The plausible mechanisms for this transformation are proposed in Scheme 3. The photosensitizer (PC+) absorbs visible light under the irradiation of 8W blue LEDs to generate its excited state (*PC+) which could oxidize TMSN3 2 to the azido radical A, which is then trapped by alkenyl silanes 1 to form B and the reduced species (PC*).[11] Sub-sequently, PC* could be oxidized by dioxygen to provide superoxide and regenerate the photocatalyst (PC+). The radical B which then reacts with superoxide and TMS+ to afford the peroxide C. Then peroxide C is then eventually converted into the carbonyl product through migration of the silyl group to the internal oxygen atom. Finally, desiliconization of intermediate D produces the desired product 3.[12]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In summary, a convenient method for the construction of α-azidoketones has been developed. The conditions are mild, and a variety of alkenyl silanes participate efficiently in the reaction. The reaction could be easily carried out in gram scale. A variety of radical sources, including trifluoromethyl radical, thiocyanate radical, bromide radical, chlorine radical, were all effectively applicable in the sustainable reaction system. The products are easily derivatized further. Current efforts in our laboratory are underway to explore various reactions of alkenyl silanes.
4 Experimental section
4.1 General Information
MeCN, N,N-dimethylformamide (DMF), and CH2Cl2 were distilled from CaH2 prior to use. 8 W blue LEDs were used as light source. The Acr+$\mathrm{MesClO}_{4}^{-}$ was prepared according to the literature.[13] NMR spectra were recorded on a Bruker-400 instrument or a Wuhan Zhongke-NiuJin- 400 instrument. 1H NMR chemical shifts were referenced to tetramethylsilane signal (δ 0). 13C NMR chemical shifts were referenced to the solvent resonance (δ 77.00, CDCl3). IR spectra were recorded on a Perkin-Elmer Spectrum One FTIR spectrometer with diamond ATR accessory. High- resolution mass spectra (HRMS) were recorded on a Waters XEVOG2-S ESI-TOF mass spectrometer. Melting points were obtained using a WRR melting point apparatus (Laboratory Devices, Shanghai Precision & Scientific Instrument Co., Ltd.).
4.2 Procedure for the synthesis of starting materials
The synthesis of 1a~1o was according to the previously reported method.[6a-6b]
4-(1-(Diphenylsilyl)vinyl)benzyl 2-((3-(trifluorometh- yl)phenyl)amino)nicotinate (1o): White solid, m.p. 91.6~94.7 ℃ [V(petroleum ether, PE)∶V(ethyl acetate, EA)=10∶1]. 1H NMR (400 MHz, CDCl3) δ: 10.33 (s, 1H), 8.40 (dd, J=4.4, 1.6 Hz, 1H), 8.28 (dd, J=8.0, 2.0 Hz, 1H), 8.08 (s, 1H), 7.86 (d, J=8.0 Hz, 1H), 7.60~7.53 (m, 4H), 7.45~7.31 (m, 11H), 7.29~7.26 (m, 1H), 6.76 (dd, J=4.8, 2.8 Hz, 1H), 6.30 (d, J=1.2 Hz, 1H), 5.71 (d, J=2.4 Hz, 1H), 5.39 (s, 1H), 5.32 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 167.2, 155.8, 153.1, 145.4, 143.3, 140.34, 140.28, 135.7, 134.1, 132.8, 132.7, 131.1 (q, JC—F=32.1 Hz), 129.9, 129.2, 128.4, 128.1, 127.0, 124.1 (q, JC—F=272.7 Hz), 123.5, 119.0 (q, JC—F=3.9 Hz), 117.1 (q, JC—F=3.9 Hz), 114.1, 107.4, 66.8; 19F NMR (376.5 MHz, CDCl3) δ: -62.6; IR (neat) ν: 2129, 1691, 1611, 1586, 1529 cm-1; HRMS (ESI) calcd for C34H28F3N2O2Si (M+ H+) 581.1867, found 581.1865.
4.3 General procedure for the synthesis of α-azido ketones
To a 25 mL overdried Schlenk flask with magnetic stirrer were added 1 (0.3 mmol, 1.0 equiv.), Acr+$\mathrm{MesClO}_{4}^{-}$ (6.2 mg, 0.015 mmol, 5 mol%), 4 Å MS (w=100% based on 1). Then, the tube was evacuated and backfilled with oxygen. Subsequently, 2 (80 μL, 2.0 equiv.) and dry MeCN (10 mL) were added by syringes. The reaction mixture was stirred under the irradiation of 8 W blue LEDs for 18 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography to afford the corresponding products 3a~3o and 2ab.
1-Azidohexan-2-one (3a): Colorless oil (26.7 mg, 63% yield). 1H NMR (400 MHz, CDCl3) δ: 3.94 (s, 2H), 2.45 (t, J=7.6 Hz, 2H), 1.65~1.56 (m, 2H), 1.39~1.28 (m, 2H), 0.92 (t, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 204.6, 57.4, 39.8, 25.5, 22.2, 13.7; IR (neat) ν: 2961, 2102, 1726, 1416, 1281 cm-1; HRMS (ESI) calcd for C6H11- N3NaO (M+Na)+ 164.0794, found 164.0794.
1-Azidoheptan-2-one (3b):[14] Colorless oil (27.5 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ: 3.93 (s, 2H), 2.44 (t, J=7.2 Hz, 2H), 1.67~1.59 (m, 2H), 1.36~1.27 (m, 4H), 0.90 (t, J=6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 204.6, 57.4, 40.0, 31.2, 23.1, 22.3, 13.8.
6-Azido-5-oxohexanenitrile (3c): Colorless oil (32.9 mg, 72% yield). 1H NMR (400 MHz, CDCl3) δ: 3.99 (s, J=6.8 Hz, 2H), 2.67 (t, J=6.8 Hz, 2H), 2.47 (t, J=6.8 Hz, 2H), 2.04~1.95 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 202.8, 118.9, 57.4, 37.7, 18.8, 16.4; IR (neat) ν: 2918, 2105, 1728, 1419, 1277 cm-1; HRMS (ESI) calcd for C6H8N4NaO (M+Na)+ 175.0590, found 175.0588.
1-Azido-4-phenylbutan-2-one (3d):[14] Colorless oil (28.4 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ: 7.32~7.26 (m, 2H), 7.24~7.15 (m, 3H), 3.86 (s, 2H), 2.95 (t, J=7.2 Hz, 2H), 2.76 (t, J=7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 203.6, 140.2, 128.6, 128.3, 126.4, 57.6, 41.6, 29.4.
4-Azido-3-oxobutyl benzoate (3e): Colorless oil (35.0 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ: 8.00 (d, J=7.6 Hz, 2H), 7.57 (t, J=7.2 Hz, 1H), 7.44 (t, J=8.0 Hz, 2H), 4.63 (t, J=6.0 Hz, 2H), 4.03 (s, 2H), 2.94 (t, J=6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 201.6, 166.3, 133.2, 129.64, 129.59, 128.4, 59.3, 57.8, 39.2; IR (neat) ν: 3002, 2105, 1719, 1602, 1454 cm-1; HRMS (ESI) calcd for C11H11N3NaO3 (M+Na)+ 256.0693, found 256.0694.
2-Azido-1-cyclohexylethan-1-one (3f):[15] Colorless oil (27.1 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ: 3.99 (s, 2H), 2.45~2.37 (m, 1H), 1.87~1.77 (m, 4H), 1.72~1.64 (m, 1H), 1.45~1.17 (m, 5H); 13C NMR (100 MHz, CDCl3) δ: 207.2, 55.8, 48.3, 28.2, 25.6, 25.4.
6-Azidodecan-5-one (3g): Colorless oil (32.0 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ: 3.78 (dd, J=8.4, 4.8 Hz, 1H), 2.54~2.48 (m, 2H), 1.82~1.74 (m, 1H), 1.71~1.63 (m, 1H), 1.62~1.55 (m, 3H), 1.43~1.29 (m, 5H), 0.95~0.90 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 207.8, 68.1, 39.3, 30.5, 27.9, 25.4, 22.3, 22.2, 13.8; IR (neat) ν: 2959, 2875, 2100, 1722, 1605, 1453 cm-1; HRMS (ESI) calcd for C10H19N3NaO (M+Na)+ 220.1420, found 220.1419.
2-Azido-1-phenylbutan-1-one (3h):[14] Colorless oil (28.3 mg, 51% yield). 1H NMR (400 MHz, CDCl3) δ: 7.94 (d, J=6.8 Hz, 2H), 7.62 (t, J=7.6 Hz, 1H), 7.50 (t, J=8.0 Hz, 2H), 4.54 (dd, J=8.4, 4.8 Hz, 1H), 2.06~1.95 (m, 1H), 1.93~1.82 (m, 1H), 1.08 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 196.7, 134.8, 133.9, 128.9, 128.5, 64.5, 24.8, 10.6.
2-Azido-1-(o-tolyl)ethan-1-one (3j):[4] Colorless oil (20.0 mg, 38% yield). 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J=7.6 Hz, 1H), 7.45 (t, J=7.6 Hz, 1H), 7.33~7.27 (m, 2H), 4.45 (s, 2H), 2.56 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 196.3, 139.5, 134.3, 132.53, 132.49, 128.3, 125.9, 56.4, 21.5.
2-Azido-1-(4-methoxyphenyl)ethan-1-one (3k):[4] White solid (31.0 mg, 54% yield), m.p. 72.8~73.5 ℃ (CDCl3) (lit.[20] 69~70 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.89 (d, J=8.8 Hz, 2H), 6.96 (d, J=8.8 Hz, 2H), 4.51 (s, 2H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 191.6, 164.2, 130.3, 127.3, 114.1, 55.5, 54.5.
2-Azido-1-(3-chloro-4-methylphenyl)ethan-1-one (3l): White solid (23.8 mg, 37% yield), m.p. 78.7~80.1 ℃ (DCM); 1H NMR (400 MHz, CDCl3) δ: 7.88 (d, J=1.2 Hz, 1H), 7.69 (dd, J=8.0,1.6 Hz, 1H), 7.36 (d, J=8.0 Hz, 1H), 4.52 (s, 2H), 2.45 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 191.8, 143.0, 135.3, 133.5, 131.4, 128.6, 126.0, 54.8, 20.5; IR (neat) ν: 2112, 1685, 1602, 1446, 1220 cm-1; HRMS (ESI) calcd for C9H8ClN3NaO (M+Na)+ 232.0248, found 232.0249.
2-Azido-1-(thiophen-3-yl)ethan-1-one (3m):[16] Colorless oil (23.1 mg, 46% yield). 1H NMR (400 MHz, CDCl3) δ: 8.11 (s, 1H), 7.55 (d, J=4.0 Hz, 1H), 7.41~7.37 (m, 1H), 4.44 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 187.6, 139.0, 132.7, 127.1, 126.5, 55.4.
2-Azido-1-(benzo[b]thiophen-5-yl)ethan-1-one (3n): Colorless oil (28.7 mg, 44% yield). 1H NMR (400 MHz, CDCl3) δ: 8.37 (d, J=1.2 Hz, 1H), 7.97 (d, J=8.8 Hz, 1H), 7.88 (dd, J=8.4, 1.2 Hz, 1H), 7.57 (d, J=5.6 Hz, 1H), 7.45 (d, J=5.6 Hz, 1H), 4.65 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 193.0, 145.2, 139.4, 130.8, 128.4, 124.5, 123.9, 123.0, 122.8, 54.9; IR (neat) ν: 2917, 2105, 1686, 1592, 1425 cm-1; HRMS (ESI) calcd for C10H7N3NaOS (M+Na)+ 240.0202, found 240.0204.
4-(2-Azidoacetyl)benzyl 2-((3-(trifluoromethyl)phenyl)- amino)nicotinate (3o): Colorless oil (46.4 mg, 34% yield). 1H NMR (400 MHz, CDCl3) δ: 10.27 (s, 1H), 8.43 (dd, J=4.4,1.6 Hz, 1H), 8.32 (dd, J=8.0, 6.0 Hz, 1H), 8.06 (s, 1H), 7.95 (d, J=8.4 Hz, 2H), 7.87 (d, J=8.4 Hz, 1H), 7.57 (d, J=8.4 Hz, 2H), 7.43 (t, J=8.0 Hz, 1H), 7.29 (d, J=7.6 Hz, 1H), 6.81 (dd, J=7.6, 4.8 Hz, 1H), 5.44 (s, 2H), 4.56 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 192.7, 167.0, 155.8, 153.5, 141.8, 140.2, 140.1, 134.2, 131.0 (q, JC—F=32.2 Hz), 129.7, 128.3, 128.2, 124.1 (q, JC—F=273.0 Hz), 123.6, 119.1 (q, JC—F=3.7 Hz), 117.1 (q, JC—F=3.7 Hz), 114.1, 106.8, 86.0, 54.8; 19F NMR (376.5 MHz, CDCl3) δ: -62.6; IR (neat) ν: 2104, 1692, 1584, 1529, 1445 cm-1; HRMS (ESI) calcd for C22H17F3N5O3 (M+H)+ 456.1278, found 456.1276.
2-Azido-1-phenylethan-1-one (2ab):[4] Colorless oil (20.8 mg, 43% yield). 1H NMR (400 MHz, CDCl3) δ: 7.91 (d, J=7.60 Hz, 2H), 7.63 (t, J=7.60 Hz, 1H), 7.51 (t, J=7.60 Hz, 2H), 4.57 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 193.2, 134.4, 134.1, 129.0, 127.9, 54.9.
4.4 Control experiment
Prepared according to the general procedure. To a 25 mL overdried Schlenk flask with magnetic stirrer was added 1a (0.3 mmol, 80.1 mg), Acr+$\mathrm{MesClO}_{4}^{-}$ (6.2 mg, 0.015 mmol, 5 mol%), 4 Å MS (79.5 mg), and TEMPO (93.8 mg, 2 equiv.). Then, the tube was evacuated and backfilled with oxygen. Subsequently, 2 (80 μL, 2.0 equiv.) and dry MeCN (10 mL) were added by syringes. After 18 h, the reaction mixture was worked up, 2% yield of 3a was monitored by 1H NMR analysis.
4.5 Scale-up experiment
Prepared according to the general procedure. To a 500 mL overdried three-necked flask with a magnetic stirrer was added 1a (3.1977 g, 12 mmol, 1.0 equiv.), Acr+- $\mathrm{MesClO}_{4}^{-}$ (0.2477 g, 0.015 mmol, 5 mol%), 4 Å MS (3.2005 g). Then, the three-necked flask was evacuated and backfilled with oxygen. Subsequently, TMSN3 (3.15 mL, 2.0 equiv.) and dry MeCN (240.0 mL) was added by syringes. The reaction mixture was stirred under the irradiation of 8 W blue LEDs for 48 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography [V(PE)∶V(EA)=10∶1] to afford 932.5 mg of 3a with Colorless oil.
Prepared according to the general procedure. To a 500 mL overdried three-necked flask with a magnetic stirrer was added 1k (3.7910 g, 12 mmol, 1.0 equiv.), Acr+$\mathrm{MesClO}_{4}^{-}$ (0.2482 g, 0.015 mmol, 5 mol%), 4 Å MS (3.802 g). Then, the three-necked flask was evacuated and backfilled with oxygen. Subsequently, TMSN3 (3.15 mL, 2.0 equiv.) and dry MeCN (240.0 mL) was added by syringes. The reaction mixture was stirred under the irradiation of 8 W blue LEDs for 48 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography [V(PE)∶V(EA)=10∶1] to afford 1.4683 g of 3k with white solid.
4.6 Synthesis of 3,3,3-trifluoro-1-(4-methoxyphenyl)- propan-1-one (4)
To a 25 mL overdried Schlenk flask with a magnetic stirrer were added 1k (0.3 mmol, 94.7 mg), Acr+- $\mathrm{MesClO}_{4}^{-}$ (6.2 mg, 0.015 mmol, 5 mol%), 4 Å MS (94.4 mg), and CF3SO2Na (187.7 mg, 4.0 equiv.). Then, the tube was evacuated and backfilled with oxygen. Subsequentl dry MeCN (10 mL) was added by syringes. Then reaction mixture was stirred under the irradiation of 8 W blue LEDs for 18 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography to afford the corresponding product. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography [V(PE)∶V(EA)=10∶1] to afford 3,3,3-trifluoro-1-(4-methoxyphe- nyl)propan-1-one (4):[17] Colorless oil (44.5 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ: 7.92 (d, J=9.2 Hz, 2H), 6.97 (d, J=8.8 Hz, 2H), 3.89 (s, 3H), 3.74 (q, J=10.0 Hz, 2H); 19F NMR: (376.5 MHz, CDCl3) δ: -61.9; 13C NMR (100 MHz, CDCl3) δ: 188.1 (q, J=2.2 Hz), 164.3, 130.8, 128.9, 125.5 (t, J=277.7 Hz), 114.1, 55.6, 41.8 (q, JC—F=28.1 Hz); 19F NMR: (376.5 MHz, CDCl3) δ: -61.9.
4.7 Synthesis of 1-phenyl-2-thiocyanatoethan-1-one (5)
To a 25 mL overdried Schlenk flask with a magnetic stirrer were added diphenyl(1-phenylvinyl)silane (0.3 mmol, 86.4 mg), Acr+$\mathrm{MesClO}_{4}^{-}$ (6.1 mg, 0.015 mmol, 5 mol%), 4 Å MS (94.4 mg), and NH4SCN (46.7 mg, 2.0 equiv.). Then, the tube was evacuated and backfilled with oxygen. Subsequently dry MeCN (10 mL) was added by syringes. Then reaction mixture was stirred under the irradiation of 8 W blue LEDs for 18 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography to afford the corresponding product. he combined filtrate was concentrated under reduced pressure and purified by flash column chromatography [V(PE)∶V(EA)=10∶1] to afford 1- phenyl-2-thiocyanatoethan-1-one (5):[18] White solid (29.1 mg, 55% yield), m.p. 72.5~73.1 ℃ (CDCl3) (lit.[21] 67.9~70.1 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.96~7.93 (m, 2H), 7.68 (t, J=7.2 Hz, 1H), 7.54 (t, J=8.0 Hz, 2H), 4.75 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 190.8, 134.8, 133.9, 129.2, 128.4, 111.8, 43.0.
4.8 Synthesis of 2-chloro-1-(4-methoxyphenyl)-eth- an-one (6)
To a 25 mL overdried Schlenk flask with a magnetic stirrer were added 1k (0.3 mmol, 94.7 mg), Acr+- $\mathrm{MesClO}_{4}^{-}$ (6.3 mg, 0.015 mmol, 5 mol%), 4 Å MS (97.3 mg). Then, the tube was evacuated and backfilled with oxygen. Subsequently dry MeOH (0.8 mL) and dry dichloromethane (DCM, 9.2 mL) were added by syringes. Then reaction mixture was stirred under the irradiation of 8 W blue LEDs for 18 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography to afford the corresponding product. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography [V(PE)∶V(EA)=10∶1] to afford 2-chloro-1-(4- methoxyphenyl)ethan-1-one (6):[19] White solid (32.6 mg, 59% yield), m.p. 101.6~102.9 ℃ (CDCl3) (lit.[22] 98~100 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.95 (d, J=9.2 Hz, 2H), 6.97 (d, J=8.8 Hz, 2H), 4.66 (s, 2H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3) 189.7, 164.2, 130.9, 127.2, 114.1, 55.6, 45.6.
4.9 Synthesis of 2-bromo-1-phenylethan-1-one (7)
To a 25 mL overdried Schlenk flask with magnetic stirrer were added diphenyl(1-phenylvinyl)silane (0.3 mmol, 86.0 mg), Acr+$\mathrm{MesClO}_{4}^{-}$ (6.3 mg, 0.015 mmol, 5 mol%), 4 Å MS (86.1 mg). Then, the tube was evacuated and backfilled with oxygen. Subsequently CHBr3 (0.8 mL) and dry MeOH (9.2 mL) were added by syringes. Then reaction mixture was stirred under the irradiation of 8 W blue LEDs for 18 h. After that the reaction was quenched by petroleum ether, filtered through a short pad of silica and eluted with ethyl acetate. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography to afford the corresponding product. The combined filtrate was concentrated under reduced pressure and purified by flash column chromatography [V(PE)∶V(EA)=10∶1] to afford 2-bromo-1-phen- ylethan-1-one (7): White solid (32.1 mg, 54% yield), m.p. 48.7~50.2 ℃ (CDCl3) (lit.[23] 49.9~52.6 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.99 (d, J=8.0 Hz, 2H), 7.61 (t, J=7.6 Hz, 1H), 7.50 (t, J=7.6 Hz, 2H), 4.46 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 191.3, 133.9, 128.9, 128.8, 30.9.
Supporting Information 1H NMR, 13C NMR spectra for the synthesized compounds 1o, 3a~3o, 2ab and 4~7. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(Cheng, F.)