


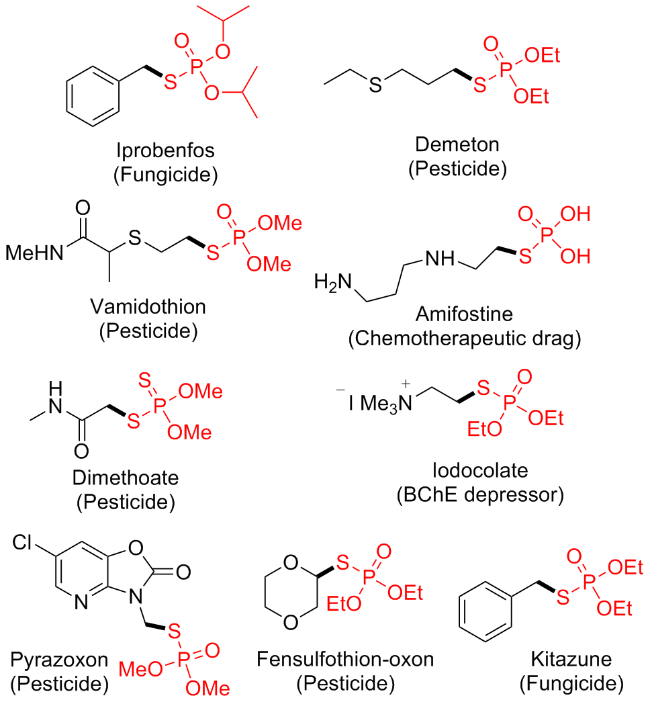
硫代磷酸酯是指含有磷硫单键的有机磷化合物, 是药物化学和合成化学中非常有价值的分子骨架, 广泛存在于许多天然产物和药物中[1]. 例如, 硫代磷酸酯作为有机磷杀虫剂在农作物保护中具有广泛的应用, 也可作为癌症化疗药物以及人类丁酰胆碱酶抑制剂[2](Figure 1). 由于硫代磷酸酯片段的独特功能, 人们在合成这些化合物方面付出了大量努力, 构建这类化合物的传统方法主要集中在P—S键形成策略上, 包括芳基硫醇与溴代磷酸酯或氯代磷酸酯的亲核取代反应[3]; 磺酰卤化物、磺酰肼、二硫化物或其它衍生物与亚磷酸酯的Michaelis-Arbuzov型反应[4], 以及芳基硫醇与P(O)H化合物的交叉脱氢偶联(CDC)[5]. 尽管这些方法做出了重大贡献, 但仍存在一些缺点, 如使用水分敏感试剂和毒性试剂、官能团耐受性低、反应条件苛刻等, 从而限制了它们的实用性. 因此, 开发方便和“绿色”的合成策略仍然是非常必要且仍是一个巨大的挑战.
2008年和2009年, Yoon, MacMillan和Stephenson团队[6]先后报道了以Ru(bpy)3Cl2作为光催化剂的不对称烷基化, [2+2]烯酮环加成和脱卤反应, 展示了可见光催化在解决合成问题方面的潜力. 自此之后, 光氧化还原催化以一种爆炸性的增长速度发展成为合成化学领域的研究热点[7]. 近年来, 可见光诱导策略合成硫代磷酸酯类化合物也得到了较大发展. 这些合成硫代磷酸酯类化合物的新型光催化反应大致可分为几类: (1)有机染料作光催化剂, (2)金属作光催化剂, (3)光催化剂和金属协同催化, (4)无光催化剂. 此外, 在过去的十年里, 电催化利用电子作为清洁的氧化剂, 从而在温和条件下提供活性中间体, 已被公认为有机合成的绿色合成工具[8]. 因此, 利用电催化合成硫代磷酸酯类化合物也受到了化学家们的广泛关注. 在此, 本综述主要介绍了近年来光/电促进的硫代磷酸酯类化合物的合成研究进展.
1 有机染料作光催化剂合成硫代磷酸酯
2016年, 张博及其合作者[9]报道了可见光诱导的硫醇1与P(O)H化合物2的氧化交叉偶联. 他们以空气作为氧化剂, 玫瑰红RB作为光催化剂, 在非常温和的条件下以中等到高的产率得到了各种S—P(O)偶联产物(Scheme 1). 在该反应中, 一系列敏感的官能团, 如羟基(3c)、氨基(3d)很容易与这种转化相容, 带有甲基(3f)、卤素(3g)的二芳基膦氧化物、烷基膦氧化物(3i)和苯基磷酸乙酯(3j)都能发生反应, 并且以中等到高的收率得到对应产物. 作者还提出了可能的反应机理: 玫瑰红RB 在可见光照射下被激发产生其激发态物质RB*, 并通过能量转移与O2相互作用产生1O2. 在这个过程中, 激发态RB*回到基态RB. 随后, 生成的1O2从硫醇1中提取一个氢原子, 得到巯基自由基4, 巯基自由基4经过偶联形成二硫醚5. 另一方面, 以P为中心的自由基6由P(O)H通过类似的氧化过程生成. 最后, 以P为中心的自由基6与巯基自由基4 (路径a)结合或与二硫键5 (路径b)反应得到产物3. 另外, 也可以通过P(O)H化合物2与二硫键5 (路径c)的亲核攻击得到产物3. 该方案是首次提出通过有机染料敏化光催化构建S—P(O)键的例子.

2018年, 吴勇及其合作者[10]报道了一种高效、绿色、实用的氧化体系, 该体系在可光催化下, P(O)H化合物9与苯硫酚8反应发生脱氢磷酸化, 以较高的产率合成了一系列硫代磷酸酯类化合物(Scheme 2). 该方法在空气条件下进行, 使用亚甲基蓝作为光催化剂, 与过渡金属光催化剂相比, 有机染料的使用特别有利, 因为它们不含金属离子, 而且价格要便宜得多. 在该反应中, 一系列含有甲氧基(10a, 10i)、卤素(10c~10e, 10h)以及敏感的氨基(10g)和羟基(10f)官能团的苯硫酚都能以良好至优异的产率得到对应的产物. 此外, 二苯基氧磷(10g~10i)、亚磷酸二甲酯(10e)也能够以高产率转化为相应的硫代磷酸酯. 该反应机理和2016年张博课题组[9]报到的机理类似.

2022年, Volla团队[11]利用伊红Y作为一种廉价和无毒的有机光催化剂, 在无金属的条件下, 通过硫酚11和三烷基磷酸酯12合成了一系列硫代磷酸酯类化合物(Scheme 3). 在该反应中, 含有卤素(13a~13c, 13f~13j)和供电子基(13d)的硫酚耐受性良好. 为了探究立体电子学对反应的影响, 作者还对各种邻取代苯硫酚进行了测试, 对应产物的产率差别不大, 说明该反应空间位阻对活性的影响较低. 此外, 各种三烷基磷酸酯(i-Pr、n-Bu、Me)也可以很好地参与反应(13h~13j). 该反应机理如下: 伊红Y在可见光照射下到达激发态形成伊红Y*, 并与苯硫酚11发生单电子转移形成伊红Y自由基阴离子和磺基自由基阳离子14, 14失去一个质子后形成磺基自由基15. 自由基15和三烷基磷酸酯12反应形成关键的磷酸基自由基中间体16, 16与过氧化物自由基进行单电子氧化形成阳离子中间体17. 17脱烷基化上的阳离子得到所需的硫代磷酸酯13. 该机理揭示了氧作为末端氧化剂的关键作用.

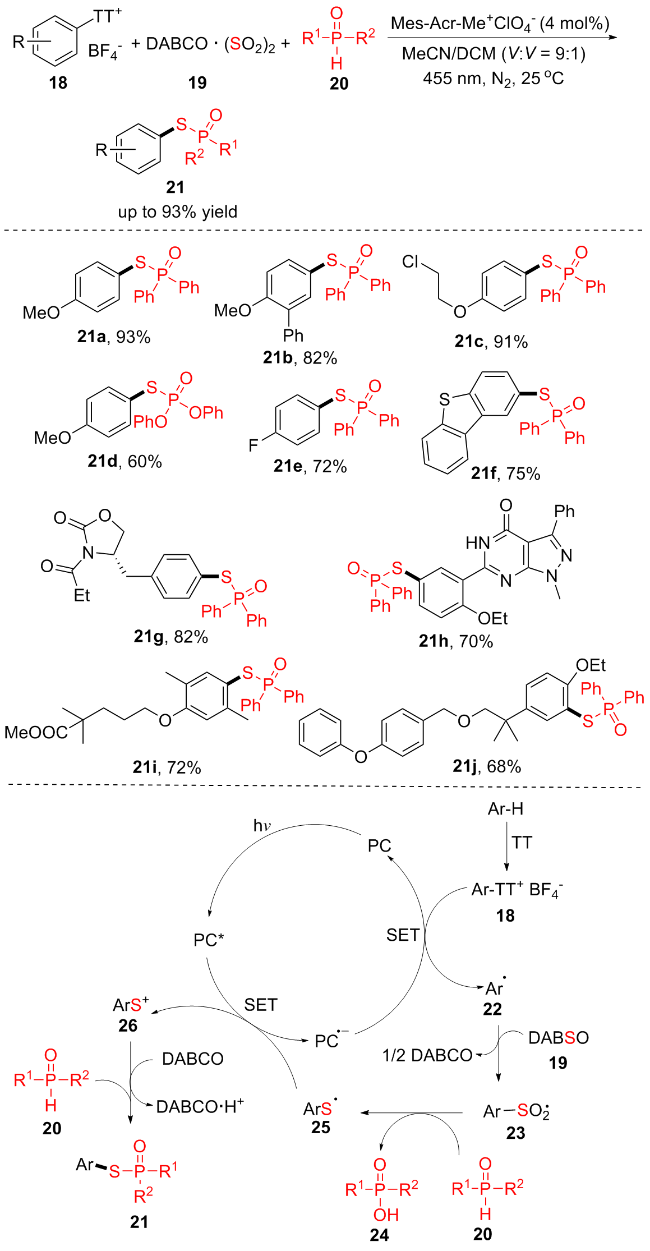
多组分反应是构建复杂分子直接有效的方法之一[12], 2024年, 金云鹤课题组[12a]报道了一种三组分合成策略, 以锍蒽盐18、双(二氧化硫)-1,4-二氮杂双环[2.2.2]辛烷加合物(DABSO) 19和二芳基膦氧化物20进行位点选择性芳烃C(sp2)—H硫代磷酸化(Scheme 4). 该方法具有显著的位点选择性, 良好的底物适用性和官能团耐受性. 此外, 单取代和多取代芳基锍盐都能够很好地参与目前的转化. 供电子基团(21a~21d)、吸电子基团(21e)和二苯并噻吩(21f)的锍蒽盐均可耐受, 二芳基磷氧化物也在该体系中表现良好(21d). 值得一提的是, 该反应进行了很多药物分子的后期修饰(21g~21j), 拓宽了构建复杂分子结构的方法, 对于合成化学和农药科学的研究具有特殊的价值. 作者还提出了可能的催化机理: 首先, PC吸收光的能量后转变为PC*, 1,4-二氮杂二环[2.2.2]辛烷(DABCO)和PC*发生单电子转移(SET)产生PC•. 随后, 锍盐18和PC•发生单电子转移生成芳基自由基22. 芳基自由基22和DABSO 19反应产生中间体23和1/2的DABCO分子. 随后, 二芳基膦氧化物20还原23得到硫自由基25. 自由基25和PC*发生单电子转移产生硫阳离子26. 最终, 26亲电攻击二芳基膦氧化物20, 在DABCO作用下脱质子后得到产物21.
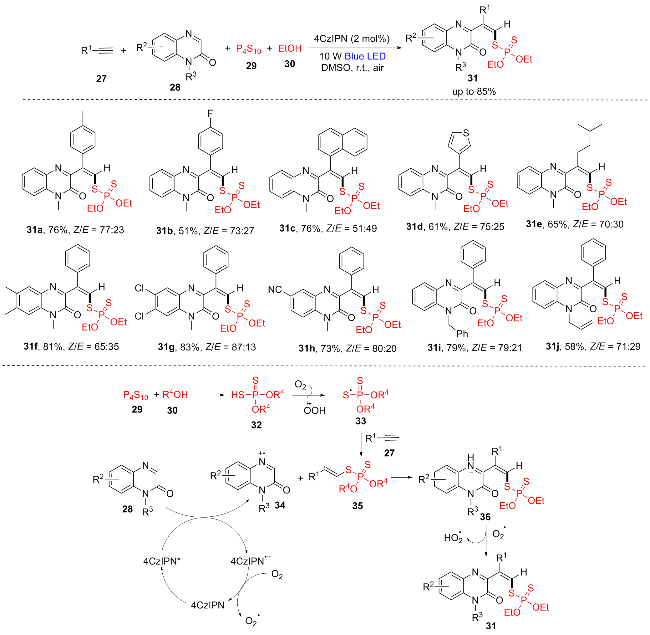
2024年, 魏伟团队[13]开发了一种无金属炔烃双官能化合成乙烯基二硫代磷酸酯的方法, 通过光氧化还原催化实现了炔烃27与喹喔啉酮28、P4S10 (29)和醇30的四组分反应(Scheme 5). 该反应仅利用4CzIPN作为光催化剂, 空气作为绿色氧化剂, 以中等到优良的产率提供了以Z异构体为主的含喹喔啉酮片段的乙烯基二硫代磷酸酯(31a~31j). 根据自由基捕获和荧光猝灭实验的结果, 他们提出了光催化自由基机理. 首先, P4S10 (29) 与30反应生成HSP(S)(OR4)2 (32), 32在空气中生成硫基自由基33, 随后33与炔烃27发生自由基加成反应生成烯基自由基中间体35, 同时在可见光照射下, 基态4CzIPN生成激发态4CzIPN*, 接着激发态4CzIPN*与喹喔啉酮28发生单电子转移(SET)生成4CzIPN•和自由基阳离子. 随后, 4CzIPN•被空气中的氧气氧化, 释放出过氧阴离子自由基, 使光催化剂再生. 此外, 自由基阳离子34与烯基自由基中间体35反应生成氮阳离子中间体36. 最后, 36通过过氧阴离子自由基去质子化生成所需产物31.
2 利用金属作为光催化剂合成硫代磷酸酯
2017年, 吴劼团队[14]报道了一种在可见光照射下, 通过四氟硼酸二芳基碘盐37、DABSO (38)和二芳基膦氧化物39的三组分反应生成硫代磷酸酯的新方法(Scheme 6). 在这一转化过程中, DABSO (38)作为硫源, 芳基磺酰自由基42的还原是实现与二芳膦氧化物的硫代化的关键步骤. 底物拓展表明该反应适用于各种吸电子基和给电子基的二芳基碘盐, 在空间位阻较大的情况下也能以良好至优异的产率得到目标化合物(40a~40h), 有取代基的二苯基氧膦也显示出适度的反应活性(40i, 40j). 他们提出了以下催化循环机理: 可见光照射下, IrIII转变为激发态IrIII*. 然后, 四氟硼酸二芳基碘盐37在激发阶段IrIII*的辅助下进行单电子转移(SET)过程, 生成芳基自由基41. 随后, 在二氧化硫中加入芳基自由基41, 得到芳基磺酰自由基42, 然后还原芳基磺酰自由基42得到芳基硫阳离子43, 加入亲核试剂39后得到相应的硫代磷酸40. 该策略为以二氧化硫为硫源合成芳基取代硫代磷酸酯衍生物提供了一种新的途径.

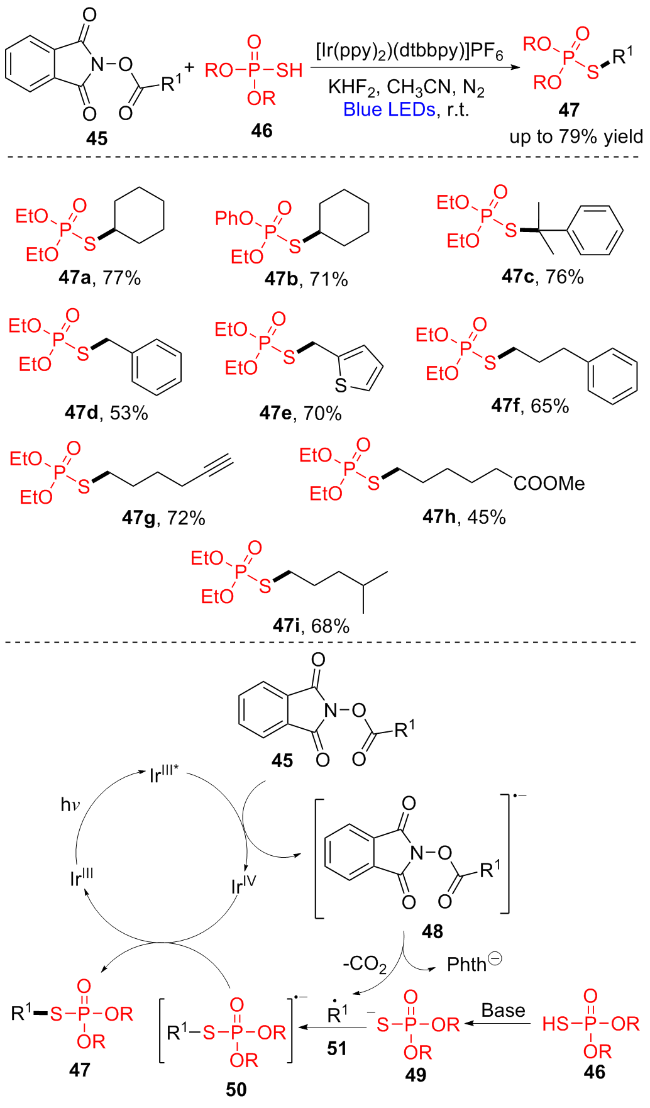
N-(酰氧基)邻苯二甲酰亚胺(NHPI酯)很容易地由羧酸合成, 最近作为烷基自由基前体被广泛研究[15]. 2021年, 宋秋玲课题组[16]报道了一种光氧化还原脱羧反应, (RO)2P(O)SH (46)作为亲核试剂与NHPI酯45发生脱羧偶联反应, 提供了一种获取硫代磷酸酯47的新方法(Scheme 7). 各种各样的羧酸, 如一级酸(47d~47i)、二级酸(47a, 47b)和三级酸(47c), 在这个脱羧反应中都可以以良好到优异的产率合成目标产物. 在标准反应条件下, 基于噻吩(47e)、末端炔烃(47g)、酯(47h)等制备的NHPI酯均被证明是相容的. 此外, 其它烷氧基取代的硫代磷酸酯也是脱羧反应的偶联底物(47b). 其反应机理如Scheme 7所示. 首先, 激发态的光催化剂IrIII*和NHPI酯45经过单电子转移(SET)形成自由基阴离子48, 然后通过脱羧和邻苯二酰亚胺阴离子产生烷基自由基51. 烷基自由基51与硫阴离子49偶联得到中间体50. 最后, IrIV物质被50还原得到目标产物47和IrIII催化剂, 完成催化循环. 该策略提供了一种从易获取的烷基羧酸出发温和而直接地制备有机硫代磷酸酯的方法.
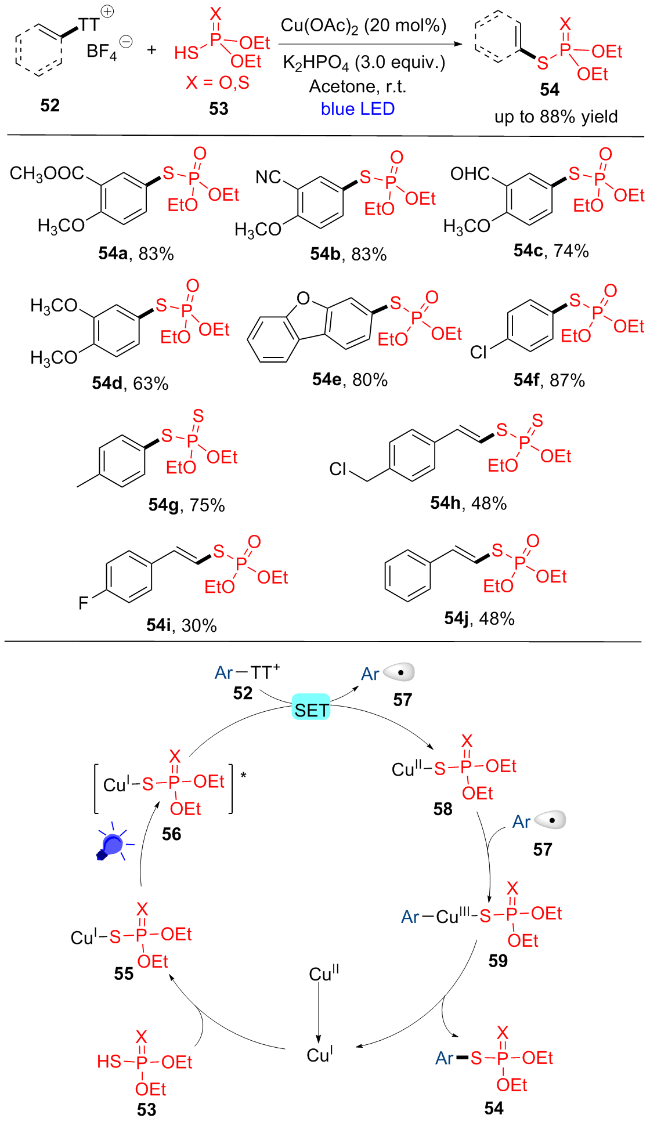
硫代磷酸酯衍生物是生物分子中显著的结构单位. 然而, 由于缺乏高效的合成方法, 阻碍了其在生物学研究中的广泛应用. 在原子经济和阶跃经济方面, 迫切需要一种比以前的方法具有更广泛的衬底的有效策略. 最近, 杨道山课题组[17]报道了可见光和铜共同催化(EtO)2- P(O)SH (53)与芳基硫鎓盐52的交叉偶联反应, 通过芳烃和烯烃的间接C—H功能化, 实现了S-芳基和S-烯基磷酸酯54的合成(Scheme 8). 该策略采用芳基和烯基硫鎓盐作为新型亲电试剂, 具有较高的区域选择性. 不同位置的吸电子或供电子基团的芳基或者烯基硫鎓盐一般具有中等到良好的产率(54a~54j), 卤素原子在此条件下可以很好地耐受(54f, 54h, 54i), 为进一步的修饰提供了机会. 一系列敏感的官能团, 如醛(54c)、氰基(54b)、酯(54a)也可很好的兼容. 值得一提的是, (RO)2- P(S)SH (54g, 54h)和杂环硫鎓盐(54e)也可以参与反应得到对应化合物. 在体系中, 一系列成功的药物分子修饰证明了该方案在生物活性分子后期功能化方面的巨大潜力. 该反应的机理是: 最初, 在磷酸氢二钾和(EtO)2P- (X)SH (53)的存在下, CuII物种被还原为CuI. CuI与(EtO)2P(X)SH (53)和磷酸氢二钾反应得到(EtO)2P(X)S- Cu(I)复合物55. 然后, 55被蓝光照射产生激发态复合物56, 随后, 芳基硫鎓盐52和一价铜物种56进行单电子转移(SET)生成芳基自由基57和CuII复合物58, 芳基自由基57与CuII复合物58反应得到CuIII复合物59. 最后, 59经过还原消除释放CuI物种和产物54. 机理研究揭示了原位形成的铜-硫配合物的关键作用, 它在可见光条件下进行了一个简单的SET过程, 从而成功地实现了自由基间的交叉偶联反应.
3 光氧化还原和金属协同催化合成硫代磷酸酯
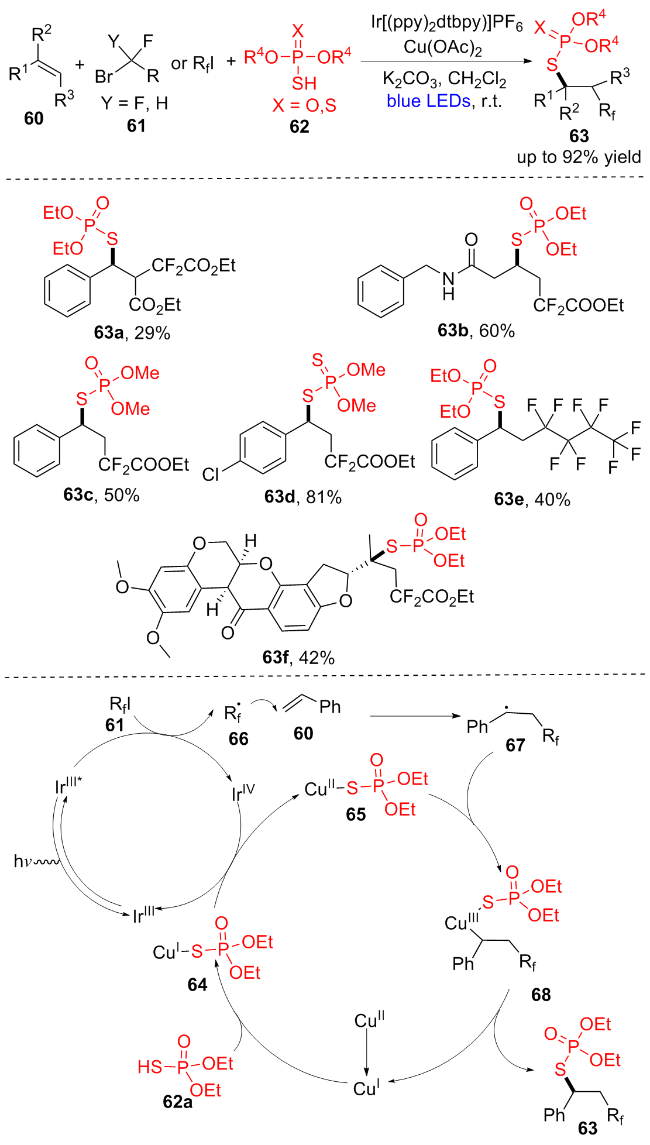
2021年, 高霞团队[18]报道了一种通过自由基接力机制的光氧化还原和铜催化氟烷基61和(R4O)2P(X)SH (62)对活化和未活化烯烃60的烯烃氟烷基硫代磷酸酯化反应(Scheme 9). 通过使用氟烷基卤化物61作为自由基前体, 并以(R4O)2P(X)SH (62)作为偶联剂, 可以在温和条件下以良好到优异的产率构建多种氟烷基取代的硫代磷酸酯(63a~63f). 该反应机理如下: 首先, 在蓝光照射下, 将IrIII激发到IrIII*, IrIII*和RfI 61发生单电子转移(SET)途径生成Rf自由基66和氧化光催化剂IrIV. 随后, Rf自由基66攻击烯烃60的C=C键, 形成烷基自由基中间体67. 同时, CuI与(EtO)2P(O)SH (62a)反应得到(EtO)2P(O)S-Cu(I)复合物64, 64被IrIV氧化生成CuII复合物65, CuII复合物65与烷基自由基67结合得到CuIII复合物68, 68通过还原消除得到最终产物63和CuI. 值得注意的是, 该反应体系可以方便地应用于生物复杂分子的后期功能化和大规模合成.
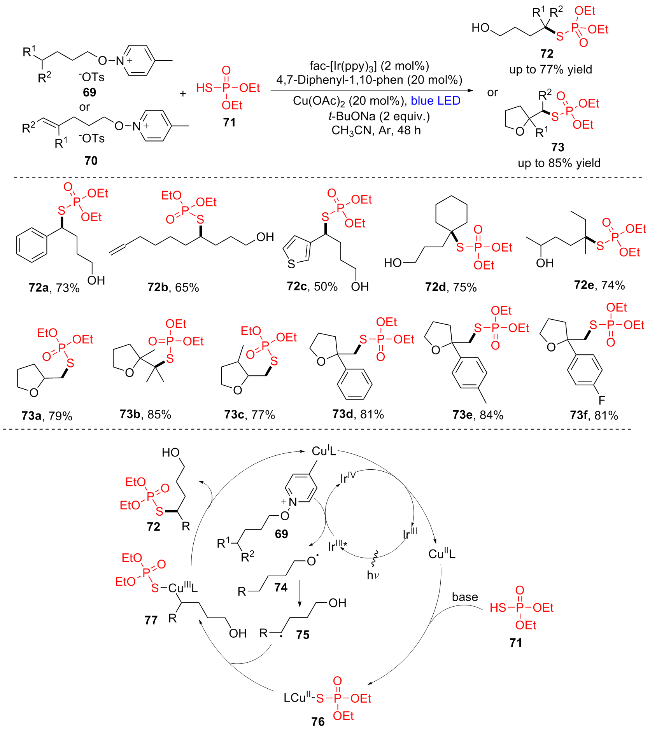
2021年, 赵玉芬团队[19]报道了一种光氧化还原/铜催化合成烷基取代硫代磷酸酯类化合物的方法, 该方法通过烷氧自由基69的1,5-氢迁移, 在光氧化还原/铜催化下δ-C(sp3)—H键与(EtO)2P(O)SH (71)偶联得到硫代磷酸酯类化合物(Scheme 10). 各种烷基、芳基和含有不稳定噻吩基和烯基的N-烷氧基吡啶盐, 经δ-C(sp3)—H硫代磷酸化以中等收率得到对应产物(72a~72e). 此外, 这一策略同样能应用于含四氢呋喃片段的烷基取代硫代磷酸酯类化合物的合成. 当起始原料为含有末端烯基的N-烷氧基吡啶盐70时, 也能以较高产率(77%~84%)获得所需的目标产物(73a~73f), 其中芳香环上具有给电子基和吸电子基的N-烷氧基吡啶盐与(EtO)2P(O)SH反应良好(73d~73f), 空间位阻对δ-C(sp3)—H硫磷化醇(73a~73c)的形成没有明显影响. 作者还提出了对应的机理: N-烷氧基吡啶盐69与激发态的IrIII*配合物发生单电子转移得到烷氧基自由基74和IrIV. IrIV与CuIL反应得到IrIII和CuIIL. 另一方面, (EtO)2P(O)SH (71)与CuIIL进行配体交换, 生成中间体(EtO)2P(O)SCuIIL (76). 烷氧基自由基74从远端分子内δ-C(sp3)—H键中提取H生成δ-C(sp3)中心自由基75, 与中间产物76结合生成CuIII配合物77. 最后, 77的还原消除产生δ-硫磷代产物72, 同时CuIL盐再生. 该反应具有较高的化学选择性和区域选择性, 将为δ-硫代磷醇和含四氢呋喃片段的烷基取代硫代磷酸酯类化合物的形成提供一种有效的方法.
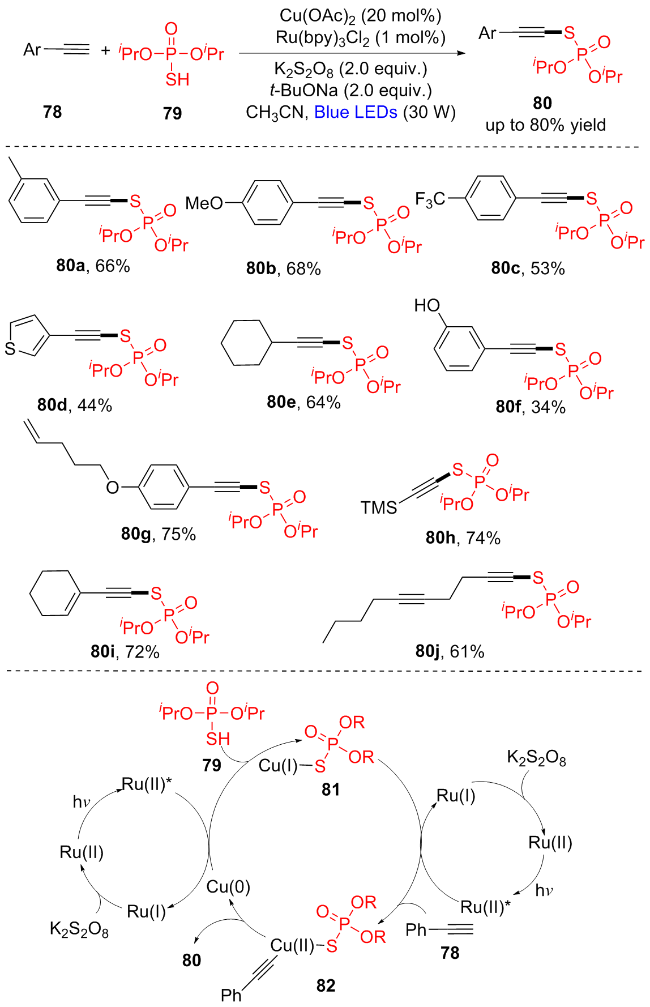
炔烃被认为是合成化学中最基本的构建模块之一, 也是合成天然产物和药物的重要中间体[20]. 从未活化的末端炔烃直接构建C(sp)—S—P(O)仍然具有很大的挑战. 2022年, 赵玉芬和唐果课题组[21]使用末端炔烃78和(iPrO)2P(O)SH (79), 通过可见光诱导的交叉脱氢偶联反应, 开创了炔基硫代磷酸酯80的第一条合成途径(Scheme 11). 该反应直接使用稳定的P(O)SH和末端炔烃, 无需制备不稳定的S—X和P—X衍生物, 避免了使用有气味的硫醇和有毒的炔溴化物, 并且官能团耐受性好. 该方法通过机理验证实验, 提出了光氧化还原/铜催化的硫代磷酸化反应的合理机制. 首先, (iPrO)2P(O)SH (79)的S—H键在碱的存在下发生初始去质子化与Cu0反应得到CuI中间体81. 其次, 在蓝光照射下, 末端炔烃78在光激发态RuII*的作用下与CuI中间体81反应形成CuII中间体82. 最后, 82还原消除产生炔硫代磷酸酯80, 同时Cu0再生, 在光激发态RuII*的作用下进行新一轮循环. 该方法在构建生物活性分子和有机磷化合物方面具有广阔的应用前景.
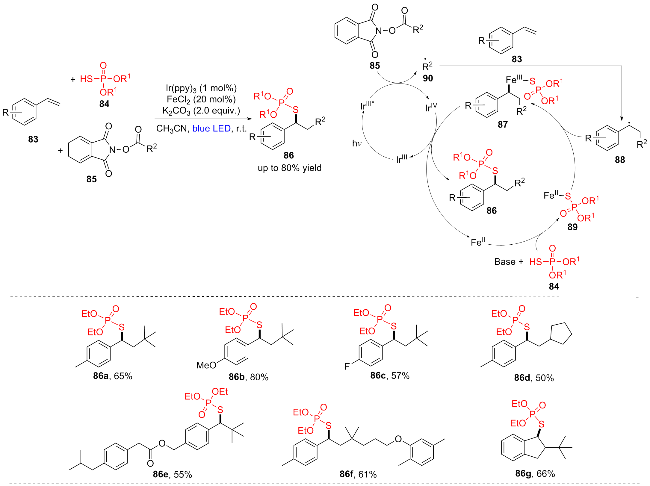
2023年, Hajra团队[22]建立了可见光和铁(II)协同催化, 以N-(酰氧基)邻苯二甲酰亚胺(NHPI酯) (85)为烷基源, 通过烯烃双官能化策略合成了一系列S-烷基硫代磷酯类化合物(Scheme 12). 具有Me, OMe, F等取代基的苯乙烯能高效地生成相应的产物(86a~86d), 非末端烯烃(86g)也能较好地参与反应, 对含有吡丙醚(86e)和吉非罗齐(86f)等生物活性分子的烯烃衍生物具有良好的耐受性, 从而为药物发现提供了潜力. 作者还进行了一系列的机理验证实验, 提出了以下机理: 在蓝色LED的存在下, IrIII被激发为IrIII*, IrIII*与NHPI酯发生单电子转移(SET), 并通过消除CO2形成烷基自由基90. 随后, 烷基自由基90攻击苯乙烯83生成亚甲基自由基中间产物88. 同时, (RO)2P(O)SH (84)在碱存在下与Fe(II)相互作用生成FeII络合物89, 89与生成的亚甲基自由基中间体88偶联生成FeIII络合物87, 最后87通过还原消除提供所需产物86, 随后将单电子转移到IrIV完成两个催化循环.
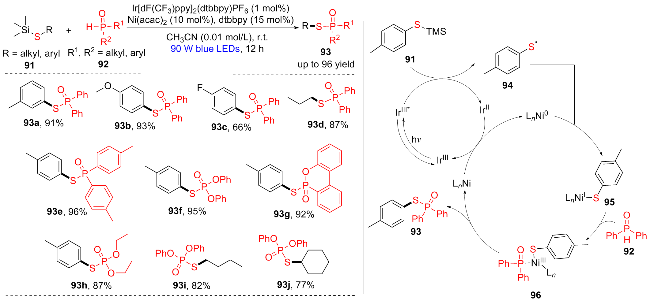
近年来, 可见光促进光氧化还原催化和镍催化结合, 在自由基介导的有机合成领域引起了较大关注. 2023年, 韩得满团队[23]报道了一种可见光促进光氧化还原和镍协同催化的策略, 用三甲基甲硅烷基硫醚91和P(O)H化合物92为原料合成硫代磷酸酯(Scheme 13). 在该方法中, 烷基和苯基取代的硅基硫醚反应活性较高(93a~93j), 苯环上带有甲基的二芳基氧化膦(93e), 9,10-二氢-9-氧杂-10-磷杂菲10-氧化物(DOPO, 93g), 乙氧基(93h)和苯氧基氧化膦(93f)都可以较好地参与偶联反应得到高产率的磷酸酯化合物. 他们提出了以下的催化循环机理: 最初, 在可见光照射下, 光激发催化剂IrIII*通过单电子转移(SET)过程氧化三甲基硅烷91, 随后脱去三甲基硅基阳离子得到关键中间体硫自由基94, LnNi0与巯基自由基94反应生成NiI中间体95, 随后与92发生氧化加成反应, 生成NiIII中间体96. NiIII中间体96进行还原消除反应生成产物93和LnNiI. 最后, IrII的单电子还原LnNiI为LnNi0, 重新启动双催化循环. 该方法为硫代磷酸酯的形成提供了直接、无毒和高效的方案, 具有优异的化学选择性和良好的官能团耐受性.
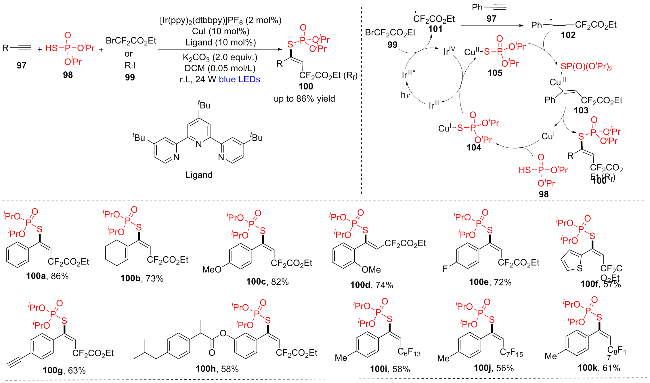
2023年, 石珊珊课题组[24]报道了一种方便的铜催化末端炔97与(iPrO)2P(O)SH (98)和氟烷基化试剂99的三组分反应, 用于合成多种具有优异区域选择性和立体选择性的(E)-β-氟烷基乙烯基硫代磷酸酯(100a~100k) (Scheme 14). 该反应有以下优点: (1)原料易得, 性质稳定; (2)反应条件温和、操作简单、官能团耐受性好(>40例); (3)易于生物活性分子的后期功能化. 该反应还进行了机理验证实验, 并且概述了炔烃的光氧化还原和铜双催化的氟烷基磷硫代反应的试验性途径. 首先, 蓝光照射IrIII会产生激发态的IrIII*, IrIII*和BrCF2COOEt 99发生单电子转移生成二氟乙酸乙酯自由基101和IrIV. 随后, 二氟乙酸乙酯基团101攻击炔烃97的碳碳叁键, 得到乙烯基加合物102. 同时, 在CuI和(iPrO)2P(O)SH (98)的相互作用之间产生(iPrO)2P(O)S-Cu(I)中间体104, 然后104被IrIV氧化产生CuII络合物105, 同时释放光催化剂IrIII, 乙烯基自由基物种102与105结合得到CuIII物种103, 经还原消除得到最终产物100和CuI催化剂.
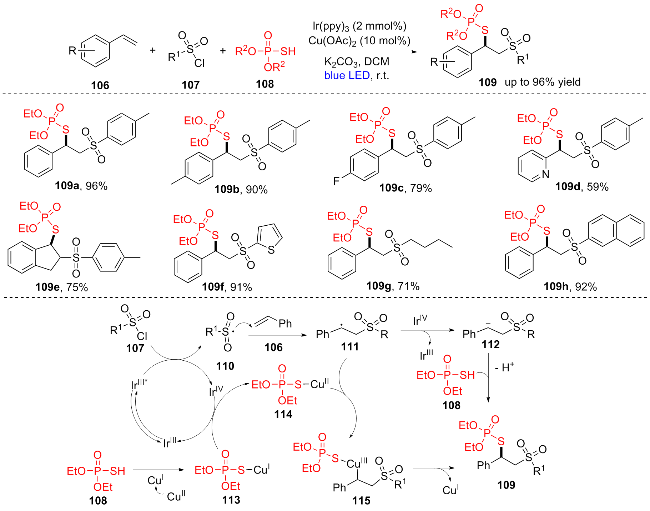
磺酰氯易于获得、稳定且具有生物相容性, 是理想的磺酰基替代品[25]. 2022年, 高霞等[26]开发了磺酰基107, 烯烃106, (R2O)2P(O)SH (108)之间的三组分自由基双官能化反应合成了一系列β-磺酰基硫代磷酸酯类化合物109 (Scheme 15). 该三组分反应具有原料易得、条件温和、产率高、底物范围广、官能团普适性广等特点, 各种取代基的末端烯烃、杂环烯烃(109d)和非末端烯烃(109e)都能以良好到优秀的产率合成对应化合物, 芳基(109a~99e)、杂环(109f)、烷基(109g)和萘(109h)取代的磺酰氯都能展现出极好的反应活性, 为β-磺酰基硫代磷酸酯的合成提供了一种高效实用的方法. 此外, 作者还提出了可能的机理: 可见光诱导IrIII产生IrIII*, 然后通过单电子转移过程还原磺酰氯107得到磺酰自由基110和氧化的IrIV. 随后, 磺酰基自由基110与烯烃106加成得到苄基自由基中间体111. 在碱的存在下, (EtO)2P- (O)SH (108)与CuI相互作用得到(EtO)2P(O)SCuI中间体113, IrIV与中间体113的反应产生CuII络合物114并再生IrIII, CuII络合物114与中间体111的结合得到CuIII络合物115, 络合物115经还原消除释放CuI, 同时得到最终产物109. 或者, 阳离子中间体112可以被亲核试剂(EtO)2P(O)SH (108)进攻得到产物109.
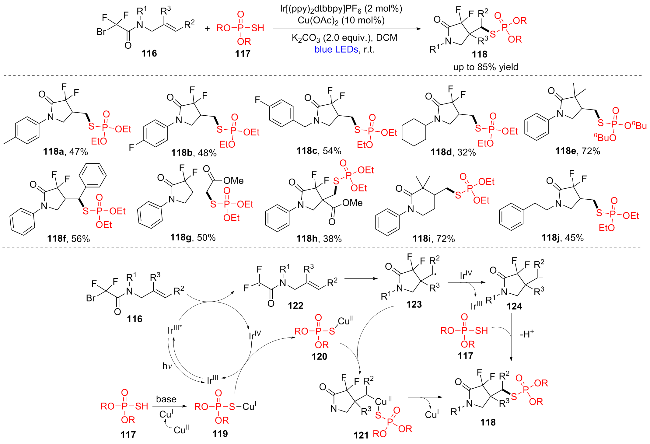
2024年, 高霞团队[27]通过N-烯丙基溴乙酰胺116的分子内自由基环化产生C(sp3)中心自由基, 进而与(RO)2P(O)SH (117)偶联制备了含吡咯烷酮片段的硫代磷酸酯类化合物(Scheme 16). 该方法可以在温和的条件下以中等至良好的产率得到目标产物(118a~118j). 作者出了可能的反应机理: 首先, 在蓝光照射下产生激发态的光催化剂IrIII*, 然后通过单电子转移(SET)过程还原116得到二氟烷基122和IrIV. 122通过分子内自由基加成得到新的自由基中间体123. 同时, (RO)2P(O)- SH (117)与CuI的相互作用形成(RO)2P(O)SCu(I)中间体119, 然后119和IrIV之间发生单电子转移得到(RO)2P- (O)S-Cu(II)配合物120和IrIII. 自由基中间体123与120反应产生CuIII物种121. 最后, 121经还原消除产生目标产物118, 并再生CuI参与新循环. 此外, 自由基中间体123可以被IrIV氧化得到阳离子中间体124, 然后与亲核(RO)2P(O)SH (117)反应得到所需产物118.
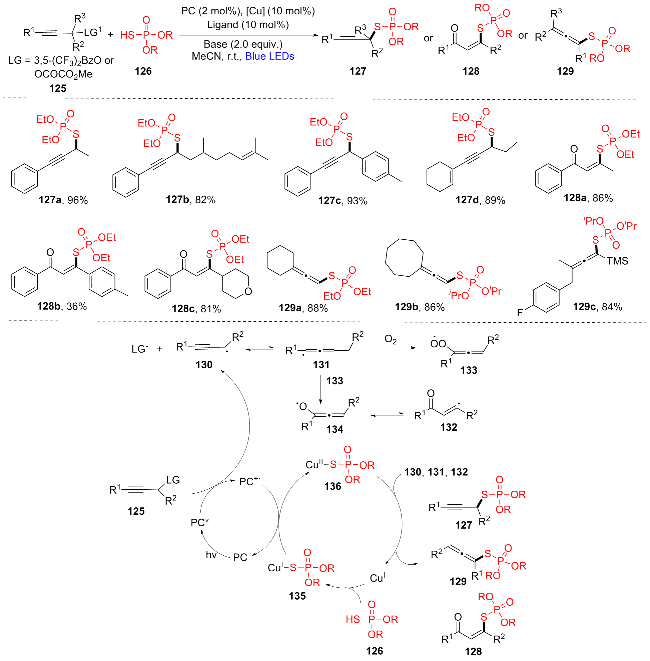
2024年, 高玉珍和石珊珊团队[28]报道了一种光氧化还原/铜双催化选择性合成硫代磷酸酯化的方法(Scheme 17). 该方法从易获取的化合物HSP(O)(OR)2 (126)对炔丙基衍生物125进行选择性硫代磷酸酯化, 提供了一种通用、温和且多功能的方法, 可从同一套简单的起始原料中选择性地合成各种S-烷基(127a~127d)、S-乙烯基(128a~128c)和S-丙二烯基硫代磷酸酯(129a~129c). 此外, 作者还提出了可能的光氧化还原和铜双催化循环机理: 在蓝光照射下炔丙基衍生物125从光催化剂(PC*)的激发态接受一个电子形成羧酸阴离子(LG)和炔丙基自由基130, 130发生自由基转移生成三取代的丙二烯基自由基131. 在空气中, 氧气对中间体131进行亲核进攻, 生成过氧自由基133, 随后131与133相互作用生成自由基134及其互变结构烯基自由基132. 同时, Cu(I)物质与HSP(O)(OR)2 (126)相互作用生成Cu(I)- SP(O)(OR)2中间体135, 135与氧化态PC•+发生SET过程生成Cu(II)配合物136, 释放出光催化剂, 完成光催化循环. 136可与自由基130、131或132偶联生成所需产物S-烷基127、S-丙二烯基128或S-乙烯基硫代磷酸酯129, Cu(I)物质再生, 完成Cu催化循环.
4 无光催化剂合成硫代磷酸酯
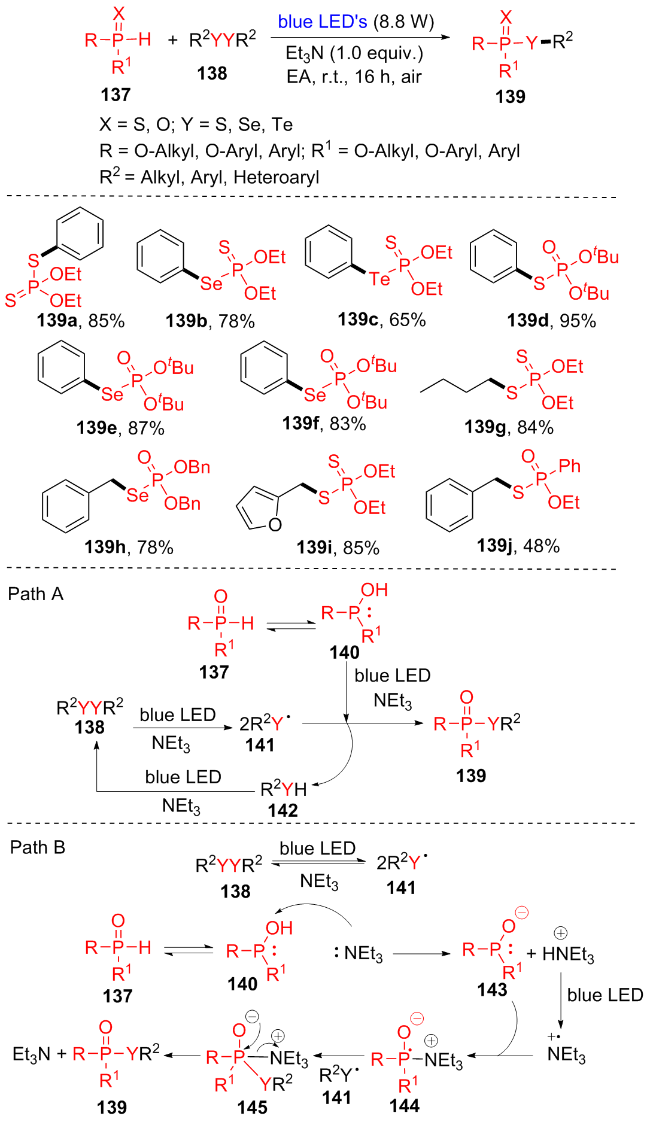
2022年, Lee课题组[29]发展了一种环境友好的模块化合成方案, 用于在蓝光发光二极管照射下利用RR1P(X)H (137)和醚138合成各种磷酸酯类化合物(Scheme 18). 蓝色LED促进的P—S、P—Se和P—Te键结构是在无金属、无配体、无氧化剂和无光催化剂的条件下形成的, 产生的化学废物少, 原子经济性高. 该方法合成的磷酸酯种类丰富, 烷基、芳基和杂环的醚都能与各种P(X)H反应, 从而以良好到优异的产率生成硫代磷酸酯、硒代磷酸酯和锑代磷酸酯(139a~139j). 这个方案能够以实际可行的产率合成农用化学品杀虫剂. 此外, 经过2,2,6,6-四甲基哌啶氧化物(TEMPO)等机理验证实验, 提出了可能的反应机理. 途径A: 通过自由基途径形成P—S键. 在碱和蓝色LED的作用下, 二硫化物138离解成巯基自由基141; 同时, P中心自由基140由亲核的P(OH)生成. 此外, 在碱和蓝光LED下, 巯基自由基141和P中心自由基140发生自由基偶联, 生成所需的目标产物139. 途径B: P—S键形成机制. P中心自由基140由亲核的P(OH)生成, Et3N和140反应得到不稳定的P中心自由基143和HEt3N+, HEt3N+在蓝光下生成Et3N+•, 随后143与Et3N+•结合得到144, 144进一步与巯基自由基141偶联得到145, 然后消除Et3N得到所需产物139.
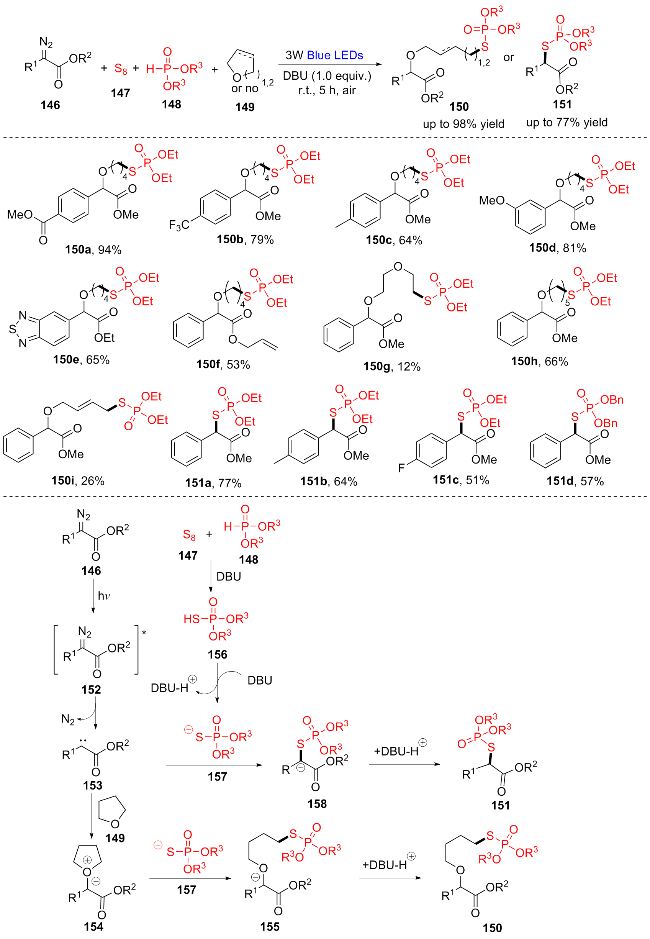
2022年, 魏伟团队[30]报道了一种α-重氮酯146、环醚149、单质硫147和(RO)2P(O)H (148)在可见光驱动下的四组分反应, 发展了新的无金属合成S-烷基硫代磷酸酯150的方法(Scheme 19). 该策略简单地利用1 equiv.的1,8-二氮杂双环十二烷(DBU)作为添加剂, 在没有任何光催化剂或过渡金属的情况下, 构建一系列收率中等至良好的S-烷基硫代酸酯(150a~150i). 值得注意的是, 如果加入2.5 equiv.的DBU作反应溶剂, α-重氮酯146、单质硫147和(RO)2P(O)H (148)三组分偶联产物也可以方便地以中高收率得到151a~151d. 底物拓展研究表明: 酯基(150a)、三氟甲基(150b)、烯基(150f)、甲氧基(150d)、氟(151c)的官能团在反应中具有良好的耐受性. 此外, 杂环重氮酯(150e)也是这种光诱导的无光催化剂四组分反应的合适底物. 该反应可能的机理如下: α-重氮酯146在可见光照射下光解生成卡宾中间体153, 并释放出N2. 同时, 元素硫147与(R3O)2P(O)H (148)相互作用得到(R3O)2P(O)SH (156), 156被DBU进一步去质子化产生硫阴离子157. 然后, 硫阴离子157将捕获卡宾中间体153, 在1,4-二氧六环中形成中间体阴离子158. 随后158质子化产生硫代磷酸烷基酯151. 或者, 当四氢呋喃(THF)用作反应介质时, 卡宾中间体153会被THF迅速捕获, 得到氧鎓叶立德中间体154. 硫阴离子157对中间体154的亲核进攻产生了中间体155. 最后, 中间体155的快速质子化将得到所需的产物150. 该反应的优点有: 温和的无金属条件、广泛的底物范围、绿色能源、良好的官能团耐受性, 满足了绿色和可持续合成化学的要求.
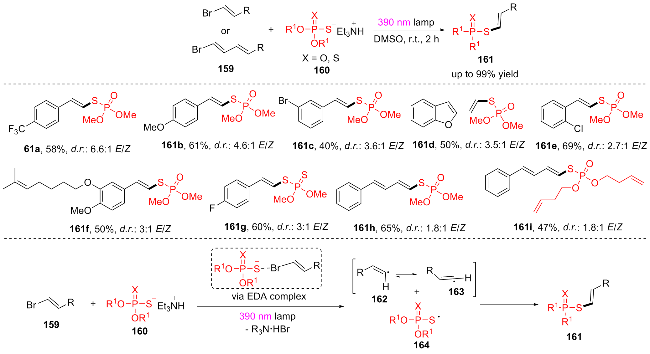
近年来, 卤素键电子供体-受体(EDA)络合物的光化学活化引起了广泛关注, 因为它可以生成碳中心自由基, 从而促进新型有机转化的发生[31]. 2023年, Plaza团队[32]提出了一种简便、通用的新型S-烯基溴和二烯基溴159合成硫代磷酸酯161的方法(Scheme 20), 该过程依赖于在390 nm紫光照射下形成的烯基或二烯基溴159(电子受体)与磷(二)硫代盐160(电子供体)之间的EDA配合物的光化学激发, 发生分子内电荷转移/碳卤键断裂产生烯基自由基162、163和巯基自由基164, 随后通过自由基偶联途径得到所需的产物. 该反应有较好的官能团耐受性, 各种吸电子和给电子的烯基溴和二烯基溴都能发生反应(161a~161h), 二丁烯基硫代磷酸酯(161i)也可以以47%的产率得到对应化合物.
5 电催化合成硫代磷酸酯
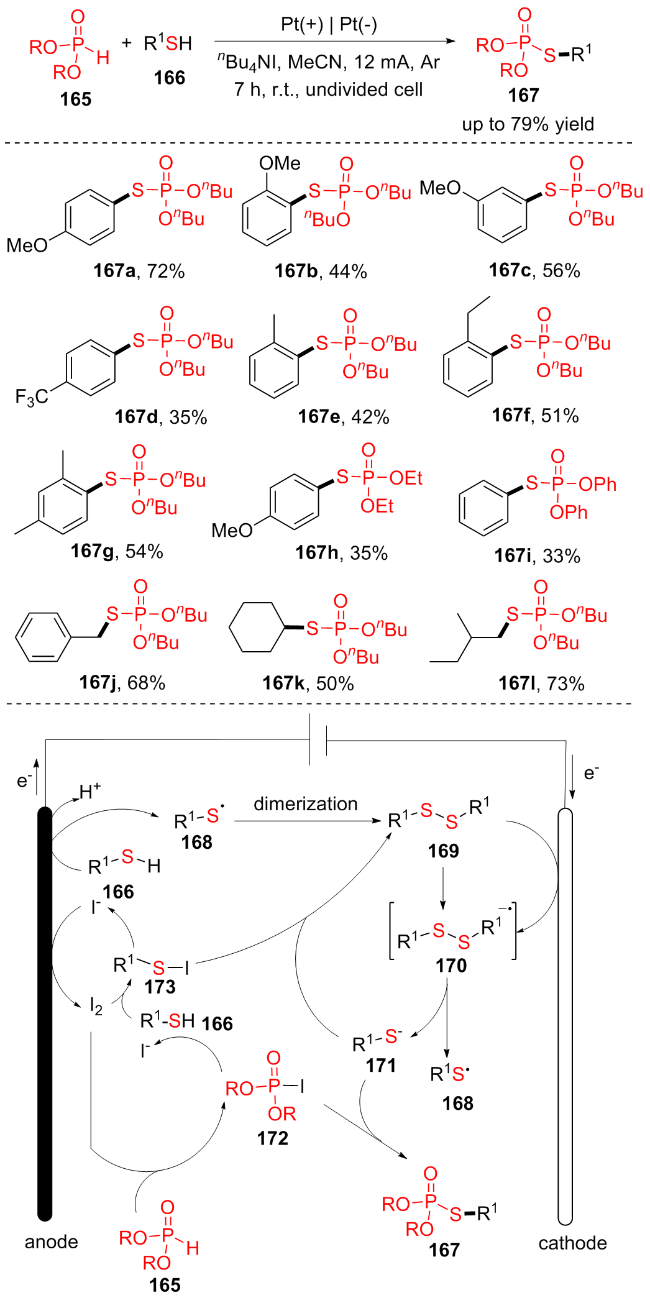
随着环保意识和可持续发展的要求, 电化学逐渐被认为是有机合成的理想工具之一. 电化学合成利用电子作为清洁的氧化还原剂, 条件温和, 原子利用率高, 符合绿色化学的特点. 迄今为止开发的形成P(O)—S键的方法中, 大多存在原子经济性和反应效率低的问题. 因此, 由P(O)—H键和S—H键引发的氧化交叉脱氢偶联(CDC)反应已成为最具吸引力的途径之一, 但是通过电化学CDC反应形成P(O)—S键尚未得到很好的证实. 2019年, Lee课题组[33]报道了在温和的反应条件下, 硫醇166与(RO)2P(O)H (165)的首次电化学氧化脱氢合成硫代磷酸酯167的新方法(Scheme 21). 该反应在室温下不加入氧化剂、过渡金属或碱的情况下进行, 具有良好的底物范围和官能团耐受性, 芳基硫醇(167a~167i)和烷基硫醇(167j~167l)与各种(RO)2P(O)H反应良好, 可以高产率地得到相应的硫代磷酸酯类化合物. 他们提出了电氧化P—H/S—H交叉偶联机理: 反应混合物中的n-Bu4NI在电化学环境下产生I2单质, I2可将(RO)2P(O)H (165)和硫醇166分别氧化为172和173. 二硫化物169是通过电化学方法从硫醇166中产生的. 二硫化物169的阴极还原生成相应的硫阴离子171和硫自由基168, 硫阴离子171与碘磷酸酯172反应生成产物. 该反应方案H2是唯一的副产物, 与传统的氧化交叉偶联方案相比, 电化学无外部氧化剂脱氢交叉偶联表现出一种绿色方法, 这可能会启发人们在更多的氧化交叉偶联反应中使用电化学方法.
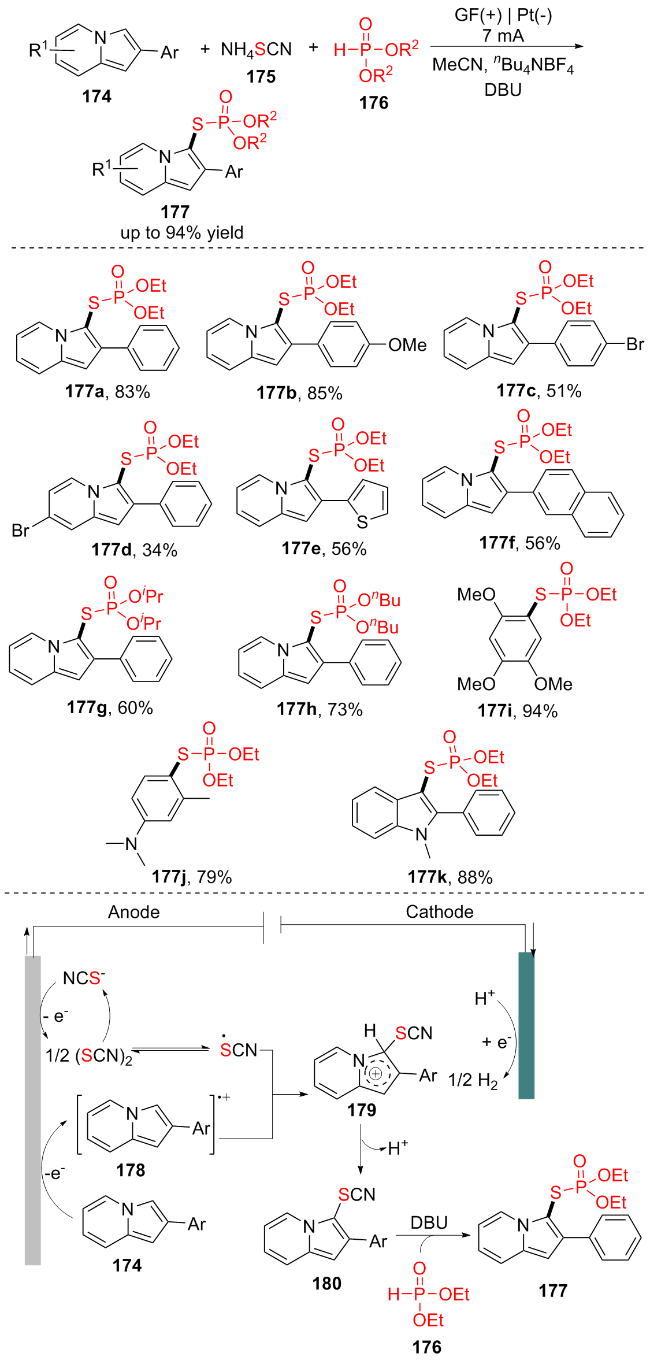
2024年, 沈振陆团队[34]报道了一种简单有效的一锅电化学氧化法, 在没有外部氧化剂和过渡金属催化剂的情况下, 实现了吲嗪的C—H硫代磷酸酯化反应. 该策略以硫氰酸盐175为硫源, (R2O)2P(O)H (176)为磷酸化试剂, 发展了一种简便实用的杂环取代硫代磷酸酯类化合物合成的新方法(Scheme 22). 此外, 此方案具有良好的底物耐受性, 所需的产品可以中等到优异的产率获得(177a~177k). 该团队还利用了循环伏安法和对照实验揭示了该反应的机理: 最初, 174在阳极表面经历单电子氧化形成阳离子自由基178, 并且NH4SCN (175)在阳极上被氧化成1/2 (SCN)2, 其可以转化成硫氰酸根. 随后, 178与硫氰酸根交叉偶联产生中间体179, 该中间体经去质子化过程产生180. 最后, 179在DBU的存在下与176反应产生所需的产物177. 同时, 氢在阴极表面释放. 该方法为室温下电化学氧化合成S-(杂)芳基硫代酸盐提供了一条绿色途径.
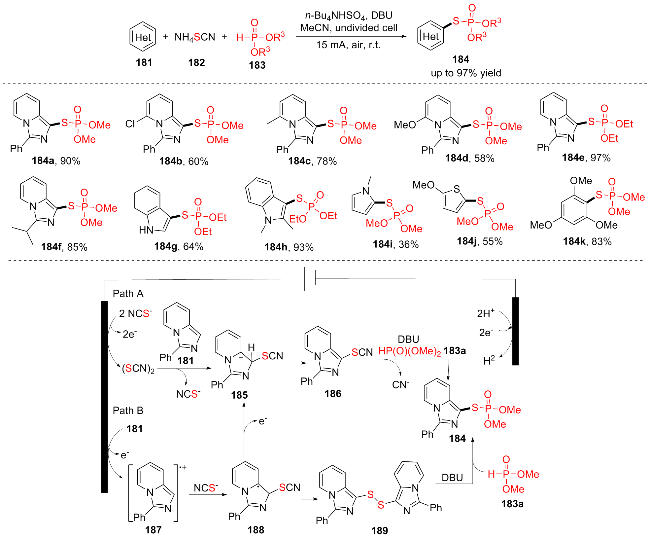
2021年, 徐坤和冯承涛团队[35]以硫氰酸盐为硫源, 在超声波辐射下发展了一种电氧化和超声协同作用的区域选择性的(杂)芳烃的C—H硫代磷酸酯化反应(Scheme 23). 该反应有以下优点: (1)底物范围广, 官能团耐受性好, 芳烃和不同取代基的咪唑并吡啶都适用于该条件; (2)不含过渡金属和外部氧化剂; (3)条件温和, 底物毒性和气味小. 遗憾的是, 具有较高氧化电位的底物在该反应中失败. 机理研究发现反应机理可能是: 最初, 咪唑[1,5-a]吡啶181经过电氧化生成自由基阳离子187, 而自由基187被NCS攻击生成自由基188, 随后自由基188进一步电氧化生成碳正离子中间体185. 或者, NCS可以在阳极被氧化, 然后发生二聚化生成亲电中间体(SCN)2. 然后(SCN)2被咪唑[1,5-a]吡啶181捕获得到碳正离子中间体185. 中间产物185随后脱质子生成硫氰化的中间产物186, 中间产物186被亲核性的(MeO)2P(O)H (183a)攻击生成产物184 (Path A), 同时中间产物188也可以转化为二硫化物189, 189与亲核(MeO)2P(O)H (183a)反应生成产物184 (Path B).
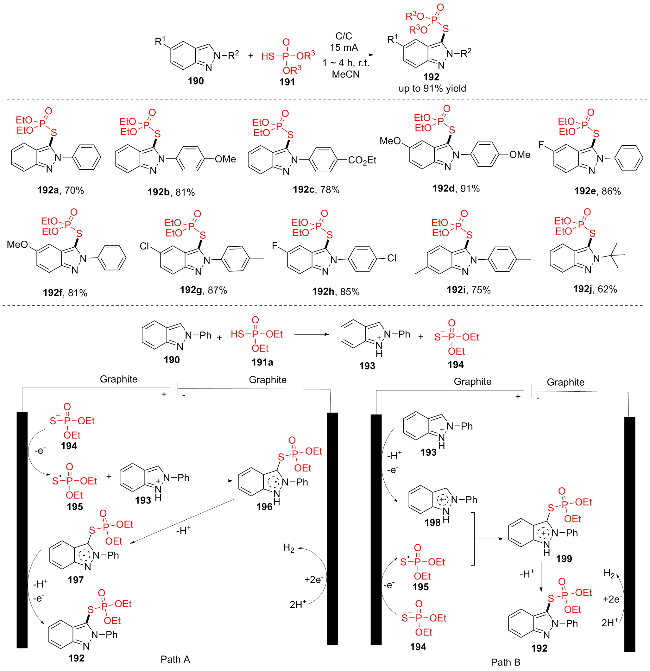
2022年, Hajra团队[36]发展了一种在无金属和无电解质条件下, 以电流为唯一氧化剂, 通过交叉偶联脱氢反应, 用(R3O)2P(O)SH (191)对2H-吲唑190进行硫代磷酸化的电化学方法(Scheme 24). 该方案对各类官能团具有较好的兼容性, 以中等到良好的产率提供目标产物(192a~192j). 该反应可能的机理如下. 途径A: 最初, 在(R3O)2P(O)SH (191)的存在下2-苯基-2-吲哚190质子化得到阳离子中间体193. (EtO)2P(O)S (194)氧化生成巯基自由基中间体195, 然后195与质子化的2-苯基-2-吲哚193偶联并形成中间体196. 接着, 通过中间体196的去质子化产生中间体197. 最后, 中间体197在阳极氧化和去质子化形成目标产物192. 此外, 氢阳离子被还原, 在阴极产生H2气体. 途径B: 193和194几乎同时在阳极被氧化, 因为两者的氧化电位非常接近, 提供阳离子自由基198和巯基自由基中间体195. 随后, 通过195和198的自由偶联形成中间体199, 随后中间体199消除质子得到最终产物192.
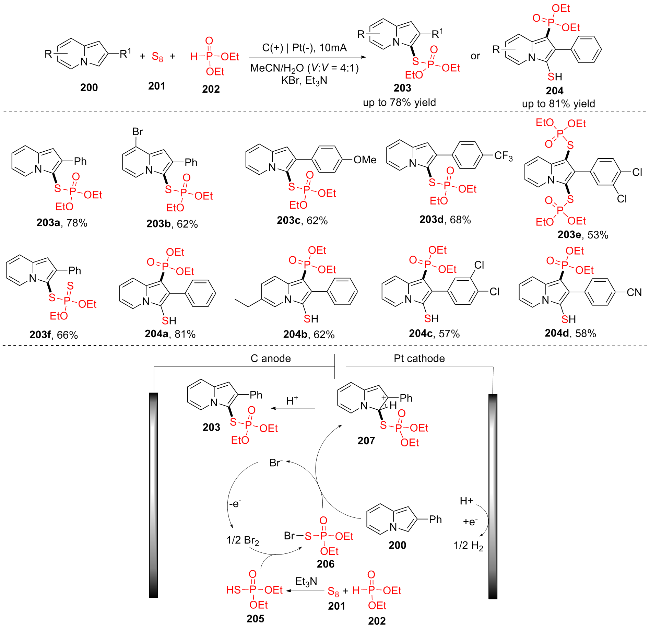
2023年, 曹华团队[37]报道了一种电化学区域选择性C—H硫代磷酸酯化反应, 在温和条件下, 通过吲嗪200、S8 (201)和(EtO)2P(O)H (202)的电化学多组分反应直接获得硫代磷酸吲嗪(Scheme 25). 这种策略利用元素硫作为无味的硫源, 为在可持续条件下形成C(sp2)— S—P键提供了新的途径, 得到了各种吸电子和给电子取代基的硫代磷酸酯(203a~203f). 作者提出了可能的反应机理: 为了形成硫代磷酸酯类产物203, 首先在阳极通过电化学氧化产生Br2. S8 (201)和(EtO)2P(O)H (202)原位反应产生(EtO)2P(O)SH (205). 随后, Br2与(EtO)2P(O)SH (205)反应得到活性物种206. 接下来, 用2-苯基吲嗪200攻击206, 得到加合物207, 接着去质子化, 得到硫代磷酸产物203.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 结论与展望
本综述对近年来利用光/电合成硫代磷酸酯类化合物的合成方法进行了归纳总结, 包括无金属光催化、金属光催化、染料光催化、光氧化还原和金属协同催化的新型光催化反应以及无外部金属和催化剂的电氧化反应. 在上述合成策略中, 光催化和电催化为S-P(O)类化合物的构建提供了更加温和、绿色的反应条件, 能够有效解决传统反应所面临的反应所需温度高、底物范围窄、原料有毒和气味大和使用外部氧化剂等问题, 为各种硫代磷酸酯的合成提供了简单而重要的补充. 芳烃 C—H键硫代磷酸酯化是构建这类化合物的有效途径, 但是光催化领域研究较少, 在电催化领域主要集中在吲嗪类杂环, 因此发展新的催化体系实现多样性杂环的 C—H硫代磷酸酯化还有较大的研究空间. 此外, 这些合成策略在工业中实现大规模生产还存在一定的困难, 有待于合成化学家们的进一步研究.
(Cheng, F.)