


氧原子是有机化合物中最常见的杂原子之一. 含氧有机化合物广泛地应用于材料化学、合成化学和药物化学等领域. 氧化反应是最常见的引入氧原子的方法之一. 然而, 传统的氧化方法如过氧化物氧化、高价金属氧化、高价碘化合物氧化等, 常因原子经济性差、成本较高、选择性差、产生大量污染物等问题而受到限制. 因此, 开发具有高选择性和高反应效率的氧化体系一直是研究的热点领域之一[1,2].
C—H键, 尤其是C(sp3)—H键, 在有机物中广泛存在, 对C(sp3)—H键的直接氧化为合成含氧有机化合物提供了高效、绿色的途径. C—H键的直接官能团化由于反应路径短, 有利于提高原子经济性和环境友好性, 已成为近几十年来的一大热点领域. 基于过渡金属催化的C—H官能团化已经得到了大力发展, 成为合成化学中一条高效的途径[3-7].
氢原子转移(hydrogen atom transfer, HAT)是一种常见的使C(sp3)—H键均裂的方法[8]. 在HAT过程中, 底物的氢原子转移到另一个高活性自由基氢受体上, 从而实现键的均裂, 使底物形成新的自由基以进行后续反应. 自进入21世纪以来, 关于HAT方法发展迅速, 并在光催化领域得到广泛应用. 在光激发下, 光催化剂可以介导或直接产生高活性自由基, 进而参与HAT过程, 实现对于C(sp3)—H键的官能团化.
1 过渡金属配合物光催化剂催化的HAT介导C(sp3)—H键氧化反应
过渡金属原子因其价态多样性和易于发生氧化还原反应的特性, 被广泛应用于多种化学反应的催化过程. 同时, 过渡金属配合物可以通过调控配体类型和过渡金属中心, 实现对反应性、立体选择性、区域选择性的精确调控. 自20世纪偶联反应迅速发展以来[12-13], 基于过渡金属催化剂的反应就一直是合成化学的研究热点之一. 近年来, 光催化与过渡金属催化协同的研究广泛[14]. 在可见光催化循环中, 具有特定结构的过渡金属配合物被可见光激发, 处于激发态的过渡金属配合物可以通过单电子转移(Single electron transfer, SET)、质子耦合电子转移(Proton-coupled electron transfer, PCET)、配体-金属电荷转移(Ligand-to-metal charge transfer, LMCT)、金属氢原子转移(Metal-catalyzed hydrogen atom transfer, MHAT)等途径, 产生高活性的自由基以进行后续的反应, 催化剂一般可以通过与底物反应、与氧化剂反应等途径恢复催化活性, 完成催化循环[14].
近年来, 具有光催化活性的Ru和Ir配合物的应用逐步深入, 在HAT介导的C(sp3)—H键氧化反应中得到了很多应用.
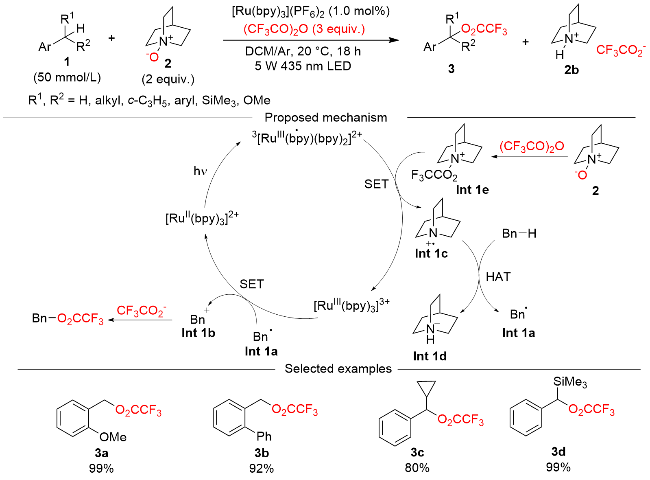
2022年, Vedernikov课题组[15]报道了一种光催化HAT介导的苄基C(sp3)—H键三氟乙酰氧基化反应, 如Scheme 1所示. 该反应使用Ru(II)配合物[Ru(bpy)3]- (PF6)2为光催化剂, 奎宁N-氧化物为HAT试剂, 三氟乙酸酐为三氟乙酰氧基化试剂, 反应在蓝光照射下进行. 该反应体系对大多数富电子的具有苄基C(sp3)—H键的底物表现出良好的适用性, 而缺电子的C(sp3)—H键无法反应. 对该反应的机理研究实验表明, 激发态的光催化剂对体系内形成的奎宁N-三氟乙酰氧化物进行单电子还原, 形成了具有HAT活性的奎宁自由基正离子中间体Int 1c. 苄基C(sp3)—H键通过HAT过程形成苄基自由基Int 1a后, 又进一步被光催化剂氧化形成苄基正离子Int 1b, 光催化剂自身被还原为Ru(II), 实现催化循环. 苄基正离子Int 1b与三氟乙酸根离子结合, 实现了苄基C(sp3)—H键的三氟乙酰氧基化.
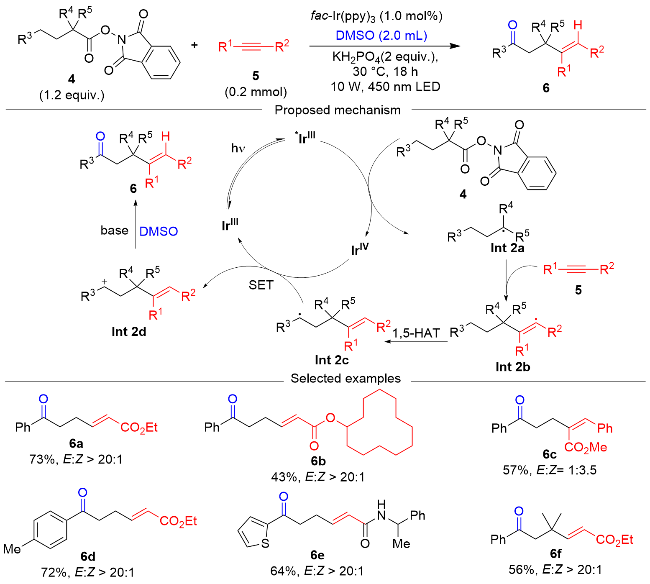
同年, 郭林和夏吾炯课题组[16]报道了一种光催化下经由烯基自由基中间体的1,5-HAT介导, 实现远端苄基C(sp3)—H键官能团化反应, 如Scheme 2所示. 在以fac-Ir(ppy)3为光催化剂、DMSO为氧化剂的条件下, 该反应可以实现远端苄位C(sp3)—H键氧化成羰基. 对该反应的底物范围探索表明, 该反应对于一系列炔烃都具有良好的适用性, 得到中等到良好的产率, 对多种N-羟基邻苯二甲酰亚胺酯(NHP esters)也具有很好的兼容性. 对该反应的机理研究表明, 在氧化反应中, 光活酯4为自由基前体, 与光活化后的催化剂发生SET过程产生烷基自由基Int 2a, 烷基自由基与炔基化合物偶联形成烯基自由基中间体Int 2b. 随后, 烯基自由基中间体Int 2b发生1,5-HAT过程形成远端苄基自由基Int 2c, 苄基自由基Int 2c与Ir(IV)反应, 被氧化为苄基正离子Int 2d, Ir(IV)被还原为Ir(III), 实现催化循环, 苄基正离子Int 2d被DMSO氧化, 实现γ-氧化.
近年来, 廉价过渡金属配合物的光催化活性也被发掘, 并成功应用于分子间HAT介导的反应. 过渡金属配合物经过LMCT过程产生Cl•、RO•、Br•、RS•等活性自由基, 这些自由基具有高HAT反应活性, 可以进行分子间的HAT过程. 近期, 基于过渡金属配合物的HAT介导光催化C(sp3)—H键氧化反应主要集中于苄基的选择性C(sp3)—H键氧化和后续反应的精确控制上.
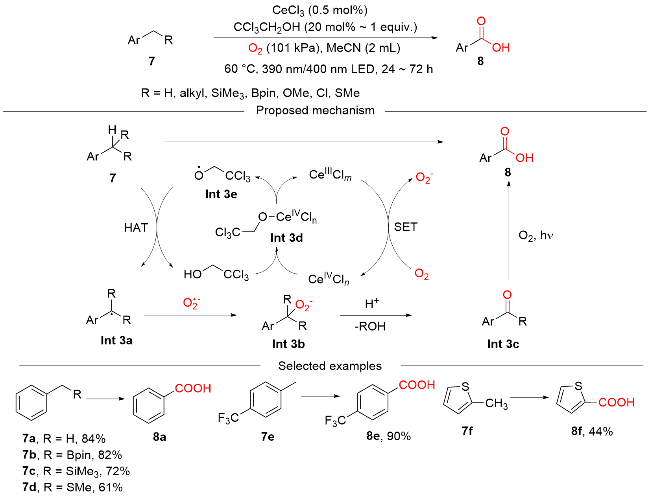
2021年, 施章杰和左智伟课题组[17]发展了一种光催化烷基芳烃的氧化反应如Scheme 3所示. 该反应使用CCl3CH2OH为HAT试剂前体, CeCl3为光催化剂, O2为氧化剂, 反应在紫光照射下进行. 底物范围探索结果表明, 该反应体系可以实现对于富电子和缺电子芳基烷烃、多取代芳基烷烃、杂环芳基烷烃的苄位氧化生成苯甲酸. 对于苄基硼化物、苄基硅化物、苄基卤化物和苄基醚等的氧化也可以顺利进行. 对该反应的机理研究表明, CeCl3被氧气氧化形成Ce(IV)化合物, 随后与CCl3CH2OH形成配合物并被紫光激发, 发生LMCT过程形成烷氧自由基Int 3e, 该中间体通过分子间HAT过程使底物生成苄基自由基Int 3a, 苄基自由基Int 3a结合超氧自由基阴离子, 实现对于苄基C(sp3)—H键的氧化. 超氧自由基阴离子来源于氧气对CeCl3的单电子氧化过程. 2022年, Walsh和Schelter课题组[18]使用[NEt4]2[CeCl6]为光催化剂和HAT试剂、空气为氧化剂, 进一步探索了Ce-Cl光催化HAT体系对C(sp3)—H键氧化反应的活性和选择性, 如Scheme 4所示. 对反应的动力学研究表明, 氧化反应的决速步是LMCT过程产生Cl•. 对底物范围的探索表明, 当使用MeCN作为溶剂时, 反应对于富电子芳基烷烃的反应性相对较好, 得到中等到良好收率的氧化产物. 光的波长会影响产物中的醇、醛酮和酸的比例, 以甲苯为模板底物, 无论使用蓝光还是紫光光源, 都只能得到产率为5%的苯甲醇产物. 使用波长为467 nm的蓝光光源时, 得到苯甲酸和苯甲醛的比例为1.0∶1; 使用紫光光源时, 苯甲酸的选择性提高, 苯甲酸与苯甲醛的比例为1.9∶1, 加入波长为330 nm的CFL光源作为额外光源以后, 苯甲酸的选择性进一步提升. 对于五元、六元和七元环烷烃, 反应可以较好地进行, 得到良好到优秀收率的氧化产物. 对于短链饱和烷烃, 该体系很难反应, 仅得到少量产物. 此外, 该体系对于氧化产物的选择性较差. 2023年, 杨义和左智伟课题组[19-21]基于之前的研究, 实现了连续流条件下蒽-铈协同催化的乙基苯类化合物苄位C(sp3)—H键选择性氧化反应[22], 如Scheme 5所示. 对乙基苯类化合物底物范围研究表明, 该体系的官能团兼容性良好, 对于富电子和缺电子乙基苯类化合物都能以良好到优秀的产率得到苯乙酮类产物, 产量为1.7~8.7 mmol/h. 蒽类化合物在这一体系中起到电子给体和受体的作用, 促进Ce催化循环的进行. 这一连续流体系的开发为Ce配合物光催化体系的工业化应用提供了思路.
图式3 CeCl3光催化HAT介导苄基C(sp3)—H键氧化反应Scheme 3 Photocatalytic HAT-mediated benzylic C(sp3)—H oxidation using CeCl3 as a photocatalyst |
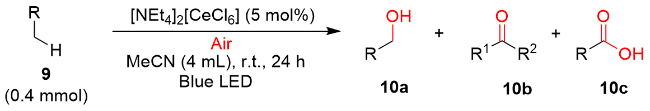
图式4 [NEt4]2[CeCl6]光催化HAT介导苄基C(sp3)—H键氧化反应Scheme 4 Photocatalytic HAT-mediated benzylic C(sp3)—H oxidation using [NEt4]2[CeCl6] as a photocatalyst |
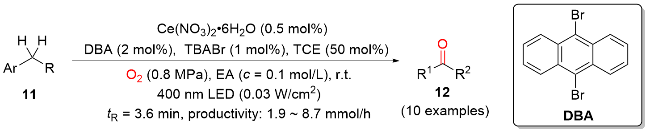
在氧化反应中, Fe配合物易通过Fenton反应生成高氧化性自由基, 这种特性虽然赋予了其高反应活性, 但也容易导致过度氧化[23-24], 因而Fe配合物在选择性氧化中的应用面临更多挑战. 近年来, 研究主要集中于苄基C(sp3)—H键的选择性氧化. [FeCl4]-配合物光激发产生氯自由基用于HAT氧化反应的报道几十年前就已经出现[25], 但是后续研究很少. 2021年, 曾荣课题组[26]和胡鹏课题组[27]分别独立发展了Fe-Cl光催化氧化断键体系. Zeng课题组使用FeCl3作为光催化剂, O2为氧化剂, 在390 nm LED灯下成功实现了对不同电性基团取代的甲基苯类化合物的氧化, 得到苯甲酸类化合物. 有苄位C(sp3)—H键的烃基苯类化合物, 如异丙苯也可以氧化断键得到苯甲酸. 同时, 当使用聚苯乙烯塑料为底物时, 该体系也可以以良好的产率得到降解氧化产物苯甲酸. Hu课题组发现在O2或空气氛围, 400 nm LED灯照射下, 多种铁盐可以催化芳烃的氧化断键或甲苯氧化, 得到苯甲酸, 其中FeCl2的效果最好. 这一体系可以实现多种类型聚苯乙烯类塑料, 包括日常生活塑料的高效降解成苯甲酸, 为聚苯乙烯类塑料的回收利用提供了新思路. 2023年, Teixeira课题组[28]发现使用FeCl3为光催化剂, O2为氧化剂, 可以实现将短链的丙烷氧化为丙酮、乙酸和异丙醇等, 进一步证明了Fe-Cl光催化氧化体系的高活性. 同年, 林军和金毅课题组[29]发展了一种铁配合物光催化的苄基C(sp3)—H键氧化酰胺化反应, 如Scheme 6所示. 该反应使用FeCl3为光催化剂和HAT试剂, 乙腈为溶剂, 甲基苯和胺为底物, 空气为氧化剂, 在紫光照射下进行. 对反应的底物范围研究结果表明, 反应体系可以适用不同电性的甲基芳烃化合物和多种脂肪胺、芳香胺, 得到中等到良好产率的氧化产物. 当芳香胺为缺电子芳胺时产率会略微下降. 当胺为邻位烯胺基取代的芳胺时, 可以生成环状的1,3-二嗪化合物. 对该反应体系的机理研究结果表明, FeCl3被紫光激发后, 通过LMCT过程产生Cl•, Cl•作为HAT试剂实现了苄基的 C—H活化, 生成苄基自由基Int 4a. 苄基自由基Int 4a与O2结合生成烷基过氧自由基Int 4b. 烷基过氧自由基Int 4b既可以作为HAT试剂, 又可以作为氧化剂实现FeCl3的催化循环, 形成苄基过氧化物Int 4c. 苄基过氧化物Int 4c脱水形成醛Int 4d, 醛Int 4d接受胺的亲核加成, 再被进一步氧化形成酰胺. 当底物胺为邻位烯胺基取代的芳胺时, Jin课题组[29]认为苄基自由基通过两种方式形成环状化合物. 第一种方式是先通过上述过程形成醛Int 4d, 醛Int 4d与胺结合形成亚胺化合物Int 4h, 再发生分子内亲核加成形成二嗪结构; 第二种方式是苄基自由基与Cl•结合生成苄氯Int 4e以后, 被氨基亲核进攻, 再进一步被氧化形成二嗪结构.
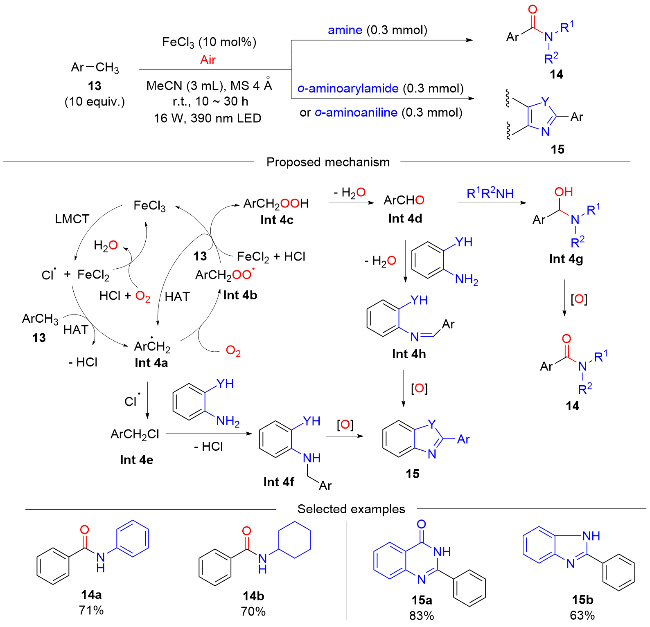
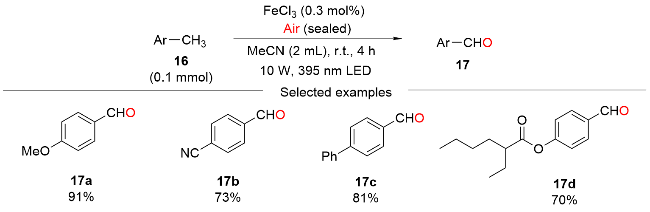
邢令宝课题组[30]在2024年发表的FeCl3光催化苄基C(sp3)—H键氧化反应体系则聚焦于将氧化产物控制到醛而不是酸, 如Scheme 7所示. 在这一工作中, FeCl3同时作为光催化剂和HAT试剂, 甲苯类化合物作为底物, 乙腈作为溶剂, 空气作为氧化剂, 反应在紫光照射下进行. 反应的机理与Jin课题组[29]的工作中的氧化过程基本一致. 敞开体系和封闭体系的对比试验表明, 对氧气的体积进行控制是反应选择性氧化到醛的关键. 底物范围探索实验结果表明, 该反应体系具有良好的官能团兼容性, 可以兼容氰基、酯基和卤素等多种基团, 对于富电子、缺电子的甲基苯化合物均能以优秀的产率和高选择性得到醛.
Cu化合物光催化氧化体系也有所发展. 2022年, 万小兵课题组[31]使用CuCl2为光催化剂, 氧气为氧化剂, 在白光照射下成功实现了HAT介导光催化芳香酰胺的氮α位C(sp3)—H键氧化反应. 当添加NH4Cl为额外氯源并使用硝基甲烷为溶剂时, 酰胺氮α位可以被选择性氧化为羰基. 当使用酸性更强的HCl为额外氯源并使用丙酮为溶剂时, 可以实现N-去甲基化反应. 该反应对于缺电子和富电子的芳香酰胺都可以顺利反应, 官能团兼容性良好.
开发新的过渡金属光催化体系也是近年来发展的一个方向. 铀酰正离子UO22+是近几年新型的一种光催化剂, 经过光激发以后, 可经由HAT过程实现对于非活化亚甲基C(sp3)—H的断裂以及经由SET过程, 实现对苯环的氧化[32-34], 但分子层面的光催化机理尚不明确. 2024年, 苏静课题组[35]探索了UO22+催化的可见光介导苄基C(sp3)—H键氧化反应的机理, 如Scheme 8所示. 理论计算和实验结果表明, UO22+被蓝光激发以后, 起到HAT试剂的作用. 整个氧化过程包括四个步骤: HAT介导的C—H键均裂、烷基自由基与三线态氧气的加成、过氧自由基和苄基自由基的偶联以及过氧键均裂. 在反应体系中先得到醇, 随后醇迅速被氧化为酮或者酸. 通过氧气对UO(OH)2+的氧化反应完成催化循环. 在选择性方面, 该反应体系会优先在较弱的C—H键发生反应. 对于不同的苄基一级醇和二级醇的底物范围探索实验表明, 该反应体系对于富电子和缺电子的芳环都能发生反应, 生成良好到优秀产率的羧酸或者酮.

十聚钨酸根离子(DT)的化学式为[W10O32]4-, 它是一种新型的光催化剂, 其结构如Scheme 9所示. 在光催化反应中, DT既可以作为HAT试剂实现C—H键的断裂, 也可以通过SET过程与底物作用[36]. 使用DT作为光催化剂催化可见光介导的C(sp3)—H键官能团化反应已经有一些报道. 2018年, Noël课题组[37]使用十聚钨酸四丁基铵(TBADT)为光催化剂, 空气为氧化剂, 在近紫外光照射下成功实现了连续流条件下的C(sp3)—H键氧化反应, 如Scheme 9所示. 底物探究结果表明该体系可以适用于苄基、烯丙基和非活化C(sp3)—H键氧化成羰基, 得到中等到良好产率的产物. 对于非活化C(sp3)— H键, 氧化反应会选择性地在亚甲基C(sp3)—H键发生. 此后, 以DT为光催化剂催化的HAT介导可见光催化C(sp3)—H键氧化反应也有一些进展. 2023年, Goddard III和Gunnoe课题组[38]使用DT为光催化剂, KCl和三氟乙酸为官能团化试剂, 在汞灯照射下成功实现了甲烷的氯化或三氟乙酰氧基化反应. 2023年, Suzuki课题组[39]使用十聚钨酸四苯基鏻(TPPDT)为光催化剂, 在近紫外光照射下实现了HAT介导的光催化酰胺氮α位 C(sp3)—H键氧化反应, 如Scheme 10所示. 对于反应的底物拓展实验证明, 该反应适用于多种芳基、环烷基酰胺, 得到少量到优秀产率的产物. 在作者提出的机理中, 光催化剂DT被光激发形成DT*, 随后通过HAT过程使底物的C(sp3)—H键发生断裂, 生成自由基Int 5a, 自由基Int 5a通过两种途径形成最终产物27. 第一种是自由基与氧气直接结合形成烷基过氧自由基Int 5b, 随后发生HAT过程形成烷基过氧化物Int 5c, 再脱水形成最终产物19; 第二种路径是自由基被单电子氧化为亚胺正离子Int 5d, 随后被H2O亲核进攻形成醇Int 5e, 醇被进一步氧化形成最终产物27.
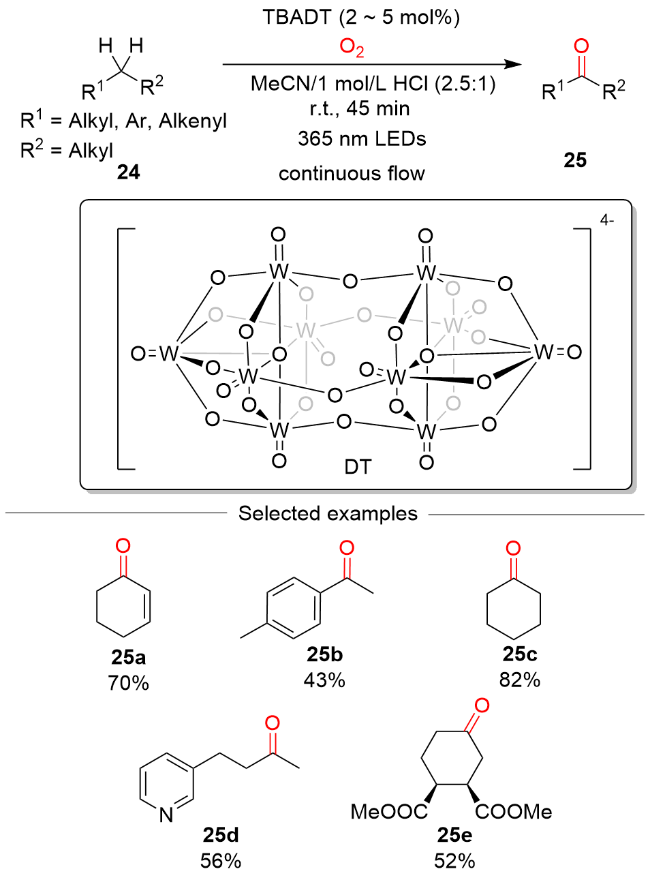
图式9 DT催化的可见光介导烯丙基、苄基和非活化 C(sp3)—H键氧化反应Scheme 9 DT-catalyzed visible light-mediated oxidation of allylic, benzylic, and unactivated C(sp3)—H bonds |
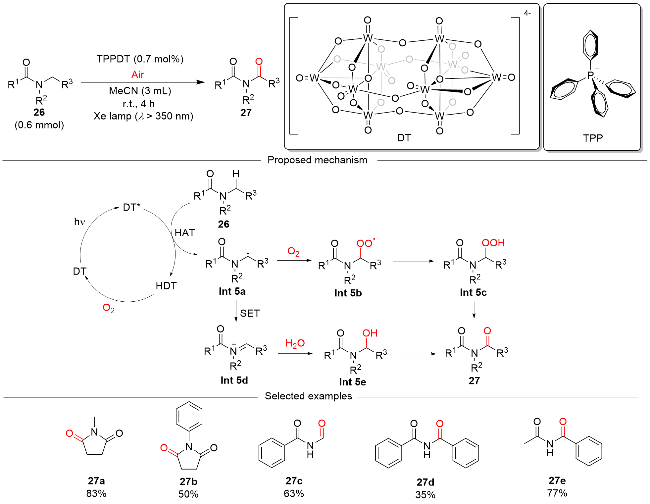
2 非金属光催化剂催化的HAT介导C(sp3)—H键氧化反应
前文提到过渡金属配合物可以实现对反应性、立体选择性和区域选择性的调控. 但过渡金属配合物也存在一些缺点, 例如产物中即便是痕量的金属残留, 也会显著影响它的生物化学和药物化学活性. 同时, 一些过渡金属元素如Pd、Os等, 本身具有高的生物毒性, 限制了这类金属配合物在生物化学、药物化学等领域的应用. 因此, 开发无金属或者无过渡金属的光催化氧化体系, 也是近年来合成化学领域研究的一大热点.
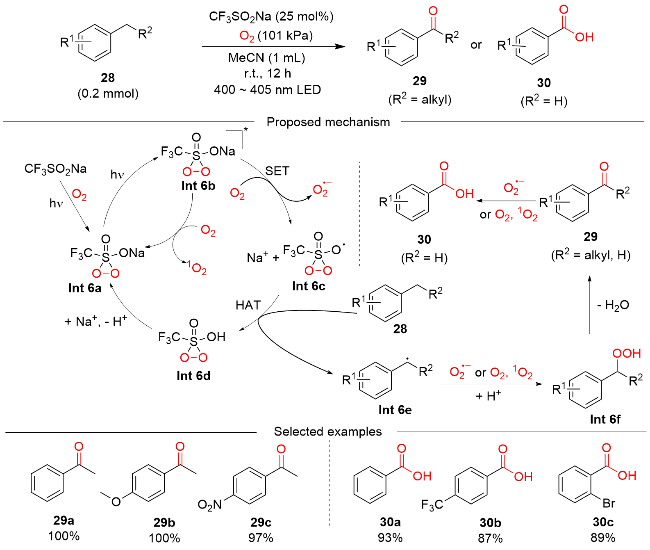
2020年, 付华课题组[40]发表了一种HAT介导的光催化苄基C(sp3)—H键氧化反应, 该反应使用O2为氧化剂, 三氟甲基亚磺酸钠为光催化剂, 在蓝光照射下进行, 可以将甲基苯类化合物和苯甲醛类化合物氧化为苯甲酸类化合物(Scheme 11). 底物范围研究实验表明, 该反应可以适用于富电子和缺电子的苯环, 得到优秀产率的产物. 机理研究实验表明, 三氟甲基亚磺酸钠与氧气在光作用下形成的具有S-O-O三元环的活性中间体Int 6a是该反应中的光催化剂和HAT试剂, 中间体被光激发以后, 进一步被氧气单电子氧化为具有HAT活性的Int 6c, 随后通过HAT过程使苄基C(sp3)—H键发生断裂生成苄基自由基Int 6e, 苄基自由基结合氧气或者活化的单线态氧形成过氧自由基, 过氧自由基通过HAT过程或者SET过程形成过氧化物Int 6f, 最后失水形成羰基, 若形成的产物是醛则会进一步氧化成酸.
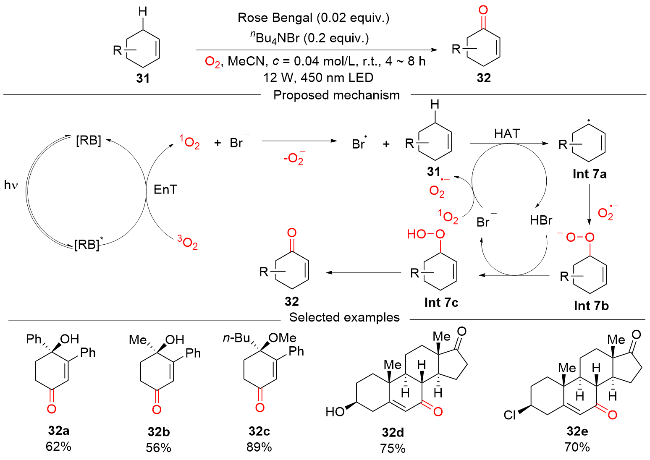
2022年, 蔡顺有课题组[41]发表了一种无金属光催化剂催化的HAT介导烯丙基C(sp3)—H键氧化体系, 如Scheme 12所示. 该体系使用氧气作为氧化剂、孟加拉玫瑰红(Rose Bengal)为光催化剂, 在蓝光照射下进行. 对底物范围的研究表明, 该实验可以适用于多种未保护三级环状烯丙醇类化合物, 得到良好产率的烯丙基C(sp3)—H键氧化产物, 且该反应体系可以应用于多种药物前体和甾族化合物的烯丙基C(sp3)—H键氧化成羰基. 对反应机理研究结果表明, 光催化剂Rose Bengal在反应中起到能量传递(Energy transfer, EnT)的作用, 在蓝光照射下, 激发态的光催化剂通过EnT过程把三线态氧转化为单线态氧, 随后单线态氧氧化Br-形成有HAT活性的Br•, Br•通过HAT过程实现烯丙位C(sp3)—H键均裂, 形成烯丙基自由基Int 7a, 烯丙基自由基与超氧自由基阴离子结合, 并结合质子形成过氧化物Int 7c, 过氧化物Int 7c脱水形成最终的氧化产物α,β-不饱和环酮28.
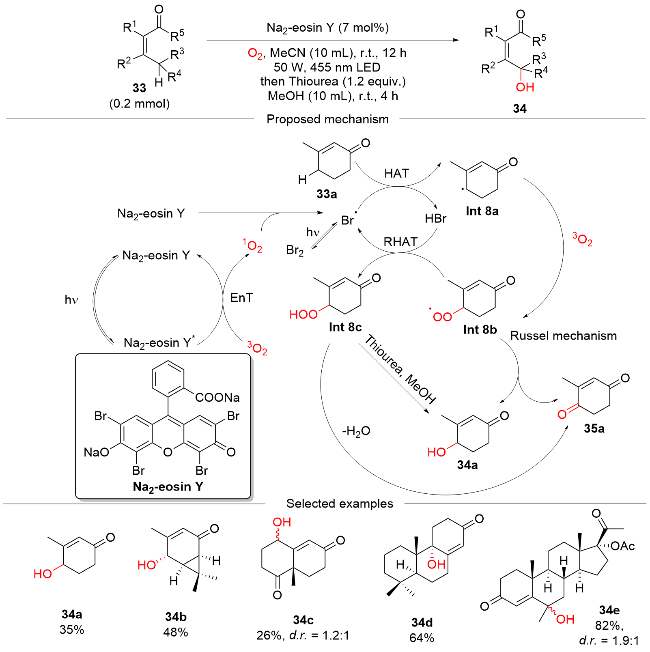
2023年, 岳建民课题组[42]使用Na2-eosin Y为光催化剂, O2为氧化剂, 先经过蓝光照射, 再使用硫脲进行还原, 成功实现了不饱和酮烯丙基C(sp3)—H键羟基化反应, 如Scheme 13所示. 这是少有的氧气氧化 C(sp3)—H键选择性羟基化反应的报道. 对于反应的条件优化实验表明, 在优化条件下, 该反应体系可以实现高选择性的不饱和酮烯丙基C(sp3)—H键羟基化, 主要产物醇和过度氧化产物酮的比例可以达到20∶1. 对反应的底物范围研究实验表明, 该反应体系的底物适用性良好, 可以用于多种倍半萜、二萜、三萜和甾族化合物等底物的烯丙位羟基化, 得到少量到优秀产率的产物. 当使用简单的六元环不饱和酮时, 反应也可以正常进行, 得到中等产率的产物. 反应的官能团兼容性良好, 可以兼容醛基、酯基、乙酰硫基等多种基团. 作者对该反应在药物后期修饰中的应用进行了探索, 结果表明该方法可以实现多种上市药物的后期羟基化修饰. 对于该反应的机理研究实验表明, 光催化剂在单线态氧作用下解离形成的Br•起到了HAT试剂的作用, 选择性对不饱和酮烯丙基C(sp3)—H键进行了拔氢. 氧化反应产生的C(sp3)—H键过氧化物Int 8c被硫脲还原, 得到羟基化产物30a. 反应中得到的羰基化副产物31a可能来源于过氧化物Int 8c的脱水, 以及过氧自由基Int 8b的Russell型歧化反应.
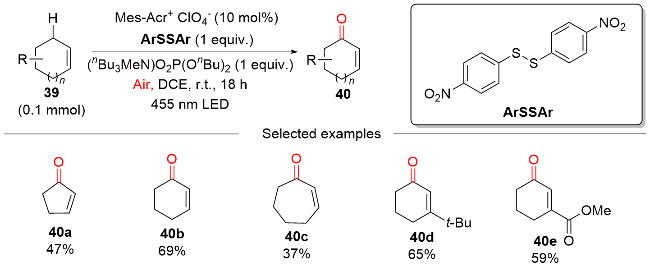
吖啶鎓盐Acr+是近几年新兴的一种光催化剂. 2024年, Yamashita和Hamashima等[43]使用Mes-Acr+ClO4-为光催化剂、Selectfluor为HAT试剂、烷氧基三甲基硅烷为甲氧基化试剂, 成功实现了苄基C(sp3)—H键的烷氧基化, 如Scheme 14所示. 该反应的底物适用性和官能团兼容性良好. 在作者提出的机理中, 光催化剂被蓝光活化以后, 通过SET过程使Selectfluor脱F-, 并生成具有HAT活性的自由基正离子Int 9a. 自由基正离子Int 9a通过HAT过程使底物形成苄基自由基Int 9b后, 苄基自由基Int 9b与光催化剂发生SET过程完成催化循环, 自身被氧化成苄基正离子Int 9c, 随后接受烷氧基的亲核进攻, 形成烷氧基化产物34. 同年, Alemán和Mancheño等[44]使用相同的光催化剂, 双(4-硝基苯基)二硫醚为HAT试剂, 空气为氧化剂, 实现了光催化HAT介导的烯丙基C(sp3)—H键氧化反应, 如Scheme 15所示. 反应在蓝光照射下进行, 并需要额外加入碱促进反应. 对于反应的底物范围研究表明, 该体系适用于环戊烯、环己烯和环庚烯的烯丙位氧化, 并且可以兼容酯基、甲氧基等基团.
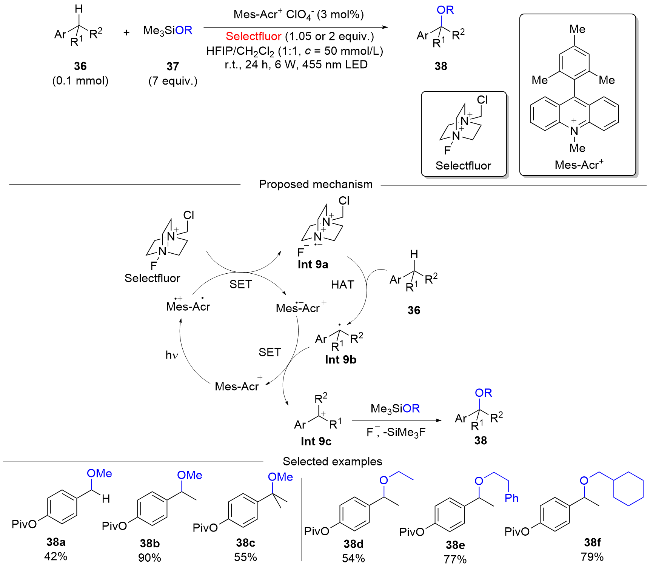
图式14 Mes-Acr+ClO4-光催化HAT介导苄基C(sp3)—H键的烷氧基化反应Scheme 14 Photocatalytic HAT-mediated alkoxylation of benzylic C(sp3)—H bonds using Mes-Acr+ClO4- as a photocatalyst |
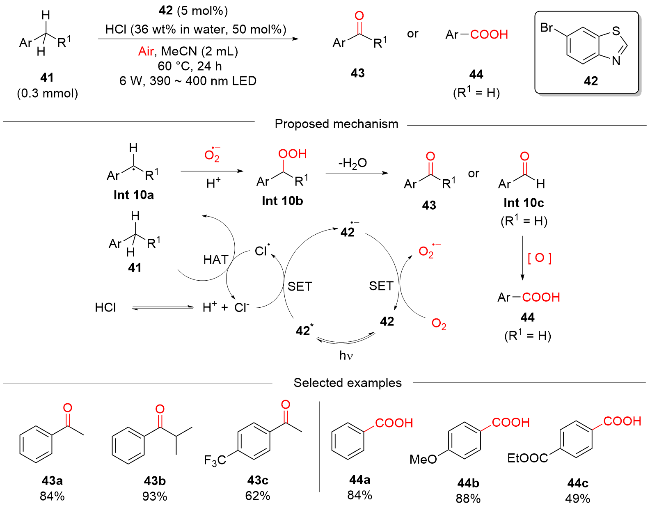
在前文中提到, 近年来过渡金属卤化物在可见光激发下通过LMCT过程产生氯自由基的报道较多, 而与此同时, 在无金属的条件下使用非金属卤化物作为HAT试剂参与光催化氧化反应的方向也备受关注. 2021年, 汪清民课题组[45]使用HCl为HAT试剂, 空气为氧化剂, 在紫光照射下实现了苄基C(sp3)—H键氧化反应, 该反应可以实现多种不同底物的苄基C(sp3)—H键氧化, 可以兼容卤原子、甲氧基、氰基、硝基等多种官能团. 2022年, 李建军课题组[46]实现了类似的可见光催化苄基C(sp3)—H键氧化反应, 该反应使用6-溴苯并[d]噻唑为光催化剂, HCl为HAT试剂, 空气为氧化剂, 反应在紫光照射下进行, 如Scheme 16所示. 对反应体系的底物范围研究实验表明, 当苄基C(sp3)—H键属于1° C—H键时, 氧化反应会直接生成酸; 属于2° C—H键时, 氧化反应停留在酮. 对于多种富电子和缺电子基团取代的苯环, 如甲氧基、酯基、氰基、卤原子和三氟甲基等, 以及部分杂原子芳环, 如噻吩环, 该反应都具有良好的兼容性, 得到中等到优秀产率的氧化产物. 机理研究表明, 光催化剂被光激发以后, 对Cl-进行单电子氧化, 产生具有HAT活性的Cl•, 随后Cl•通过HAT过程实现苄基C(sp3)—H键断裂, 将底物45转化为苄基自由基Int 10a. 苄基自由基Int 10a与超氧自由基负离子结合, 从而实现苄基C(sp3)—H键氧化. 超氧自由基来源于氧气对还原态光催化剂的单电子氧化反应.
除了非金属氯化物以外, 非金属溴化物也是常见的HAT试剂. 2022年, 刘宁课题组[47]发表了一种可见光催化HAT介导苄基C(sp3)—H键的氧化反应, 如Scheme 17所示. 该反应使用CBr4为催化剂和HAT试剂, 空气为氧化剂, 在紫光照射下进行, 将苄基C(sp3)—H键氧化到酮. 在优化实验中, 刘宁课题组发现加入四丁基高氯酸铵(tetra-butylammonium perchlorate, TBAPC)可以提高氧化反应的产率. 底物范围研究表明, 该反应可以兼容甲氧基、卤原子、硝基和氰基等多种取代基, 并且对于二苯甲烷的氧化反应可以实现克级制备. 2024年, 刘尊奇和陈建宾课题组[48]使用NH4Br为HAT试剂, 氧气为氧化剂, 氙灯为光源, 在光照和50 ℃下反应, 成功实现了光催化HAT介导苄基C(sp3)—H键氧化, 将甲基苯类化合物高选择性地氧化为苯甲酸类化合物.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 总结与展望
对近几年氢原子转移介导的光催化C(sp3)—H键氧化反应的进展进行了综述, 这部分只占氧化反应很小一部分, 是一个处于发展中的新方向. 在光催化剂方面, 越来越脱离传统的贵金属光催化体系的局限, 以Ce、Fe等廉价过渡金属元素为中心的新型光催化体系不断发展. 此外, 有机光催化剂也被越来越多地应用到此类反应, 吖啶鎓盐等新的光催化体系在光催化C(sp3)—H键氧化反应的应用也在持续推进. 在氧化剂的选择方面, 氧气的使用最为广泛, 符合现代绿色化学对氧化反应的要求. 与传统氧化反应相比, 可控的光催化条件也使反应条件更加温和, 并减少对环境有危害的化合物生成, 同时也提高了反应选择性, 拓宽了底物范围.
然而, 目前还面临很多反应发展的困境. 在底物的类型上, 主要集中于富电子苄基C(sp3)—H键和烯丙基C(sp3)—H键, 对非活化的C(sp3)—H键选择性氧化反应还缺乏手段, 而这恰恰是最有吸引力和最有应用前景的部分. 在氧化程度上, 目前依然难以控制, 主要氧化产物是酮、羧酸及羧酸衍生物, 以醛和醇为主产物的报道较少. 通过选择性氧化反应得到醇在合成化学和大宗化学品制备中有重要的应用, 但目前依旧是光催化 C(sp3)—H键氧化反应的一大难题, 当前的解决方案主要是将反应中生成的过氧化物还原为醇. 总体而言, 目前光催化HAT介导的C(sp3)—H键氧化反应还是局限于传统氧化反应的范畴, 对于氧化反应最关键的区域选择性、氧化程度和立体控制等方面还没有得到充分发展. 通过发现更多具有选择性的光催化剂、发现具有选择性氧化能力的氧化剂, 将过渡金属光催化剂与不对称配体结合运用等途径, 实现氧化反应的区域选择性、氧化程度选择性、立体化学控制, 可能是未来光催化C(sp3)—H键氧化反应的发展方向.
光催化C(sp3)—H键氧化反应的条件温和、选择性强、有可能实现多种类型的控制性氧化, 在未来, 相信光催化C(sp3)—H键氧化反应的相关研究还会不断深入和拓展, 更好服务于合成化学、药物化学和大宗化学品合成等领域.
(Lu, Y.)