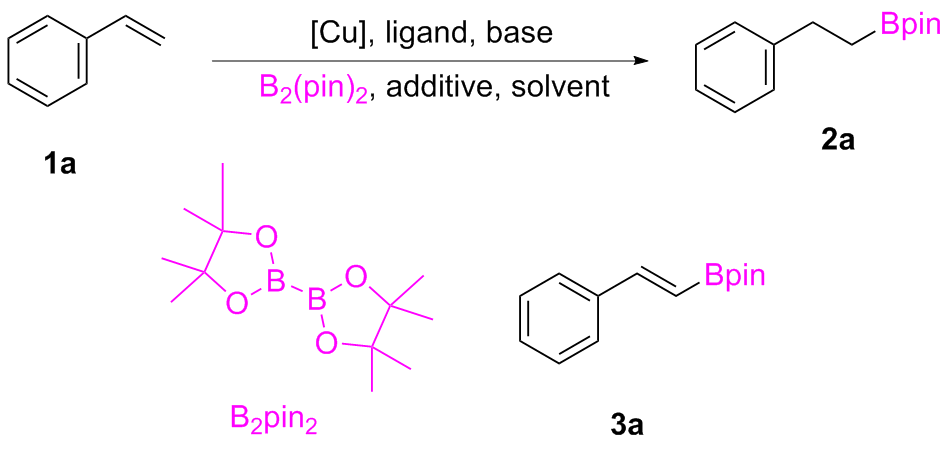
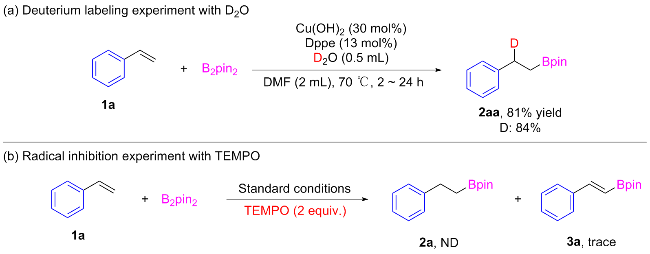
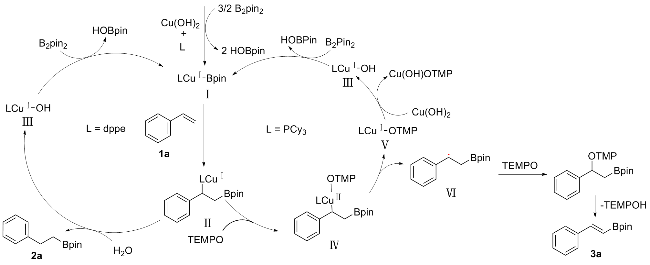
2-Phenylethylboronic acid pinacol ester (
2a) and
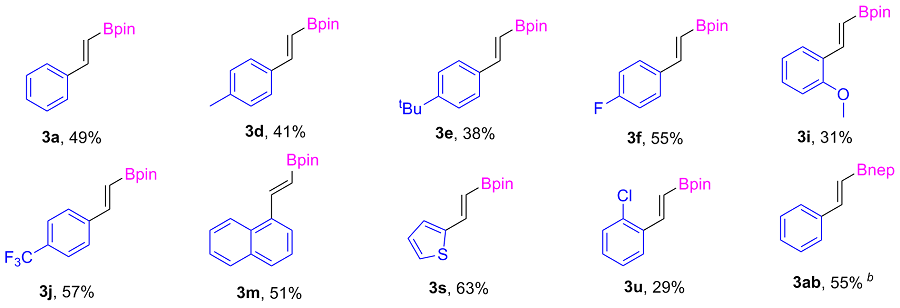
E- styrylboronic acid pinacol ester (
3a) were found when styrene (
1a) reacted with B
2pin
2 in DMF in the presence of copper organic phosphine complexes. Presence of water in reaction system provides protons that facilitate the hydrogenative borylation reaction. And the increase of amount of water improves the
regio-control of the morphology of
2a. However, the use of copper chloride instead of cuprous chloride increases the yield to 70% (
Table 1, Entries 1~3). Moreover, inorganic copper hydroxide can promote the yield to 95%. Especially, copper hydroxide acts not only as a catalyst but also as a base compared to LiO
tBu, NaOC
2H
5, K
2CO
3, NaHCO
3 and NaOH, increasing yields up to 95% (
Table 1, Entries 3~8). Among the selected solvents, DMF appears to be good in high yields. The effect of different solvents on the reaction is also enormous. DMF appears to have a high yield of 95% in the selected solvents, especially when compared to 1,4-dioxane and THF yields (
Table 1, Entries 8~10). Water has also been tried as a green solvent with a yield of only 41%~58% with the addition of co-solvents (
Table 1, Entries 11~13). DCE has only 5% yield, probably because it is not compatible with H
2O (
Table 1, Entry 14). Next, the ligands are optimized for experiments. When the ligand is Dppe or Dppbz, the yield is 95%, but using Dppbz will lead to the decrease of selectivity. When the ligand is Dppb, the yield is only 29%, due to the formation of an unstable seven-membered ring with the coordination of ligand and copper metal. The yield of the ligand is 90% for PPh
3, suggesting that the monodentate ligand may also have a high yield. When the ligand is PCy
3, the yield is 34%, which may be unfavorable for addition because of excessive cyclohexyl hindrance (
Table 1, Entries 15~18). The use of methanol as proton source results in reduced yields. In contrast, water serves as the most environmentally benign proton source, thereby negating the necessity for further screening (
Table 1, Entry 19). The absence of catalysts, ligands, or proton sources all lead to diminished reaction yields or absolute failure to form the desired products (
Table 1, Entries 20~22). At room temperature, a 24 h reaction yields 90%, suggesting that temperature may primarily influence the reaction rate (
Table 1, Entry 23). A yield of 30% is observed when using only 1 mol% catalyst and ligand (
Table 1, Entry 24). Without the addition of external base, the yield decreases to 56% (
Table 1, Entry 25). However, a 95% yield can be achieved by adding 0.3 equiv. of copper hydroxide as catalyst and base (
Table 1, Entry 26). The reaction also proceeds efficiently under atmospheric conditions and demonstrates good tolerance to oxygen (
Table 1, Entry 27). The low reactivity when using copper oxide as catalyst confirms that the active catalyst is indeed copper hydroxide Cu(OH)₂ rather than the product of thermal decomposition of Cu(OH)₂ (
Table 1, Entry 28).



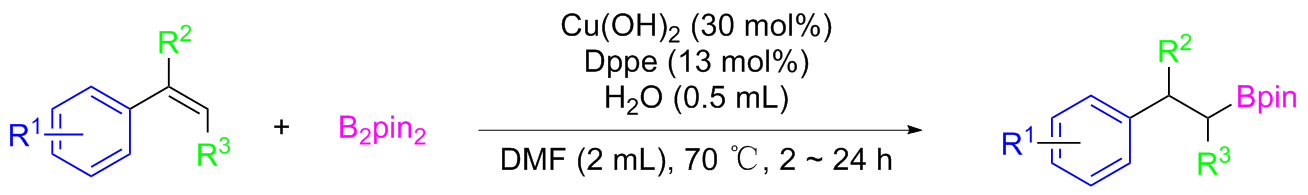


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}