


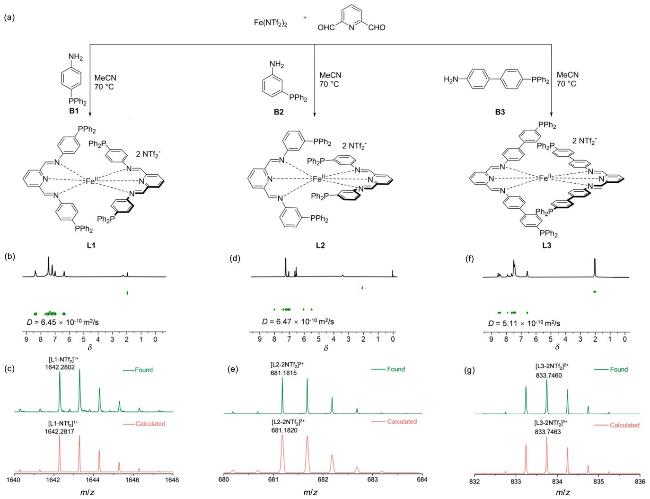
图1 (a) L1~L3的合成路线; (b) L1的扩散排序谱图; (c) L1的高分辨质谱图; (d) L2的扩散排序谱图; (e) L2的高分辨质谱图; (f) L3的扩散排序谱图; (g) L3的高分辨质谱图Figure 1 (a) Synthetic routes of L1~L3; (b) Diffusion-ordered spectroscopy (DOSY) of L1; (c) High-resolution ESI-MS spectrum of L1; (d) Diffusion-ordered spectroscopy of L2; (e) High-resolution ESI-MS spectrum of L2; (f) Diffusion-ordered spectroscopy of L3; (g) High-resolution ESI-MS spectrum of L3 |
1 结果与讨论
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
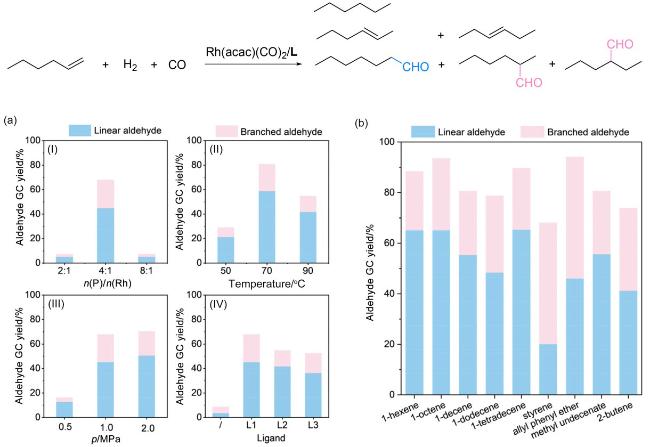
图3 (a) 1-己烯的氢甲酰化反应; (b) L1参与的不同烯烃的氢甲酰化反应Figure 3 (a) Hydroformylation reaction of 1-hexene; (b) Hydroformylation reactions of different olefins involving L1 Reaction conditions: (a) 1-hexene (4.0 mmol), Rh(acac)(CO)2 (4×10-3 mmol), H2/CO (V∶V=1∶1), toluene (3.0 mL), 5 h. (Ⅰ) 90 ℃, 1 MPa; (Ⅱ) n(P)∶n(Rh)=4∶1, 1 MPa; (Ⅲ) n(P)∶n(Rh)=4∶1, 90 ℃; (Ⅳ) n(P)∶n(Rh)=4∶1, 90 ℃, 1 MPa; (b) Substrate (4.0 mmol), n(P)∶n(Rh)=4∶1, Rh(acac)(CO)2 (4×10-3 mmol), H2/CO (V∶V=1∶1), 90 ℃, 2 MPa, toluene (3.0 mL), 12 h. |