

硅是元素周期表第四主族的成员, 是sp³杂化碳的生物电子等排体, 但两者之间也存在显著差异: (1)硅的空3d轨道使其更易形成稳定的五配位或六配位复合物[1]; (2)硅的原子半径和电正性均高于碳. 这些差异使得含硅化合物在物理化学性质上与全碳化合物有显著不同[2-6]. 例如, 将硅原子引入药物骨架后, 含硅药物相比于碳类似物, 展现出更强的细胞渗透性和更高的生物活性和更低的毒性, 从而提高药物的治疗潜力或实现药物的再利用. 因此, 将硅原子替换碳原子, 并保持分子其余部分不变的碳-硅“转换”或硅掺入, 已成为开发新型分子的创新策略之一[7-9], 并被广泛应用于新药研发和有机光电材料等领域.
四氢化萘和苯并环庚烷的结构单元广泛存在于许多上市药物和天然产物中. 通过碳-硅“转换”或硅掺入来高效合成含硅的四氢化萘和苯并环庚硅烷衍生物, 具有重要的学术和实际意义. 然而, 现有的合成方法存在诸多局限性, 如适用范围有限、反应条件苛刻以及多步骤反应复杂等, 这些因素导致含硅四氢化萘和苯并环庚硅烷的开发进展缓慢. 因此, 迫切需要一种通用且高效的合成方法来构建这类骨架[10-14].
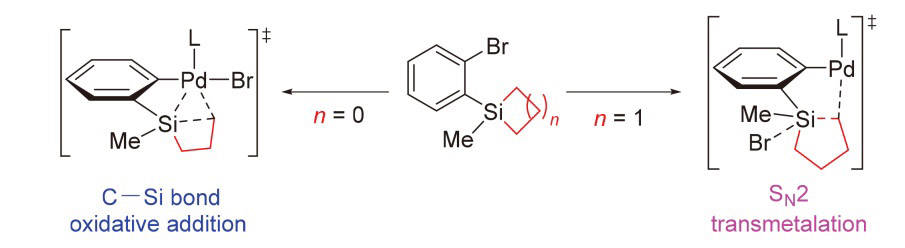
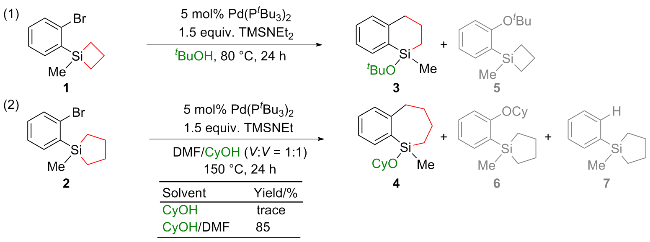
合成新型含硅分子是有机合成领域的重要课题之一[15-16], 其中硅环作为常用的硅烷化试剂, 在合成中具有独特的优势. 硅环可通过释放环张力活化C—Si键, 发生环加成或环扩张反应, 生成多种含硅化合物[17-26]. 最近, 赵东兵课题组[27]报道了在Pd催化下, 环丁硅烷和环戊硅烷通过释放环张力开环, 与邻位的芳基溴化物及醇类溶剂发生交叉偶联反应, 构建C—C键和Si—O键, 从而获得含硅的四氢化萘和苯并环庚硅烷衍生物(Scheme 1). 虽然实验结果令人鼓舞, 但仍有几个亟待解决的关键问题: (1)为什么该反应中的环戊硅烷反应温度高于环丁硅烷? (2)为什么在环戊硅烷体系中必须添加N,N-二甲基甲酰胺(DMF)溶剂才能获得产物? (3) C—O交叉偶联的副产物(如5, 6)是如何被抑制的? 基于我们对硅环开环的研究兴趣, 本工作拟采用密度泛函理论(DFT)计算, 对Scheme 1所示的两个原型反应进行详细研究, 期望通过计算结果深入理解这些反应的机制.
1 结果与讨论
1.1 环丁硅烷或环戊硅烷开环/交叉偶联反应的两种可能路径
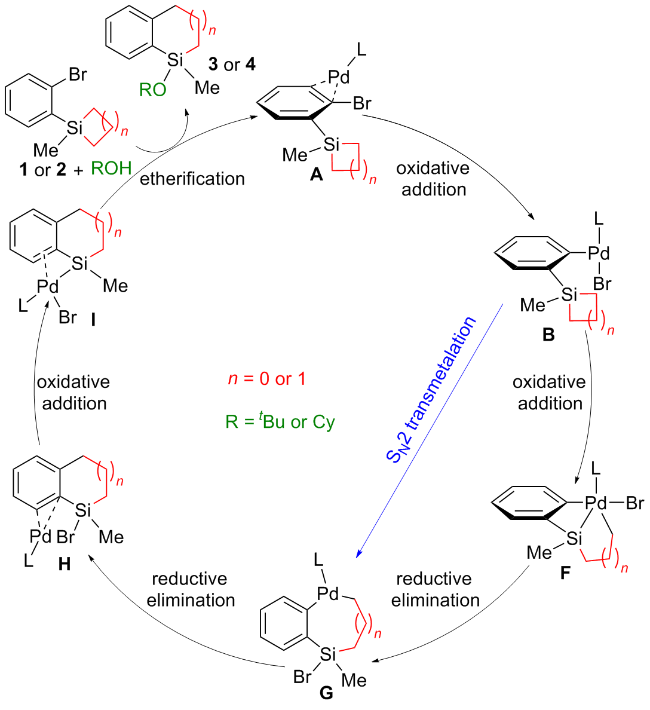
基于实验结果, 实验组认为Pd(0)催化剂A作为催化循环的起始点, 首先与底物发生C—Br键的氧化加成, 形成Pd(II)物种B. 从物种B出发, 存在两种可能的反应路径: (1)物种B中的Pd—Br键与硅环发生分子内转金属化, 生成Pd(II)物种C, 随后Si—Br键与溶剂ROH发生醚化, 生成Pd(II)物种D (Scheme 2中黑色路径). (2)物种B与ROH分子发生Br/OR配体交换, 形成Pd(II)物种E, 随后Pd—OR键与硅环发生分子内σ-键复分解, 生成Pd(II)物种D (Scheme 2中蓝色路径). 最后, Pd(II)物种D发生C—C键的还原消除释放产物. 然而, 从机理角度考虑仍存在其他可能性: (1) Pd(0)物种是否可以先与硅环反应, 再断裂C—Br键? (2)硅环是否可以通过氧化加成实现开环? (3)环丁硅烷和环戊硅烷是否经历相同的催化循环? 解答这些问题同样有助于深入理解反应的本质机制.
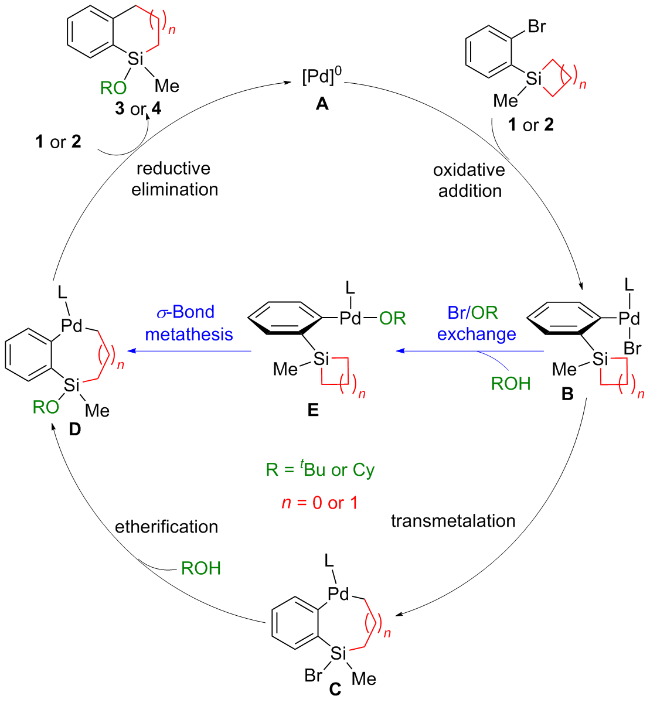
1.2 Pd催化下环丁硅烷的开环/交叉偶联反应机理
以Scheme 1中的第一个反应为模板反应, 图1展示了环丁硅烷在开环/交叉偶联反应中的热力学过程. 根据林振阳课题组[28]对钯催化C—X (Br、I)活化的理论研究, 以底物1与Pd(0)配位的复合物IM1作为势能面的相对零点. IM1通过三元环过渡态TS1进行C—Br键的氧化加成, 生成中间体IM2, 需克服能垒8.3 kcal/mol (1 cal=4.18 J). IM1还可通过过渡态TS8发生C—Si键的氧化加成释放环张力, 然而TS8的活化能比TS1高3.1 kcal/mol. 接下来, IM2可通过Y形结构的过渡态TS2 (ΔΔG≠=1.9 kcal/mol)异构化为更稳定的中间体IM3, 其中PtBu3与Br处于对位, 并放热10.1 kcal/mol.
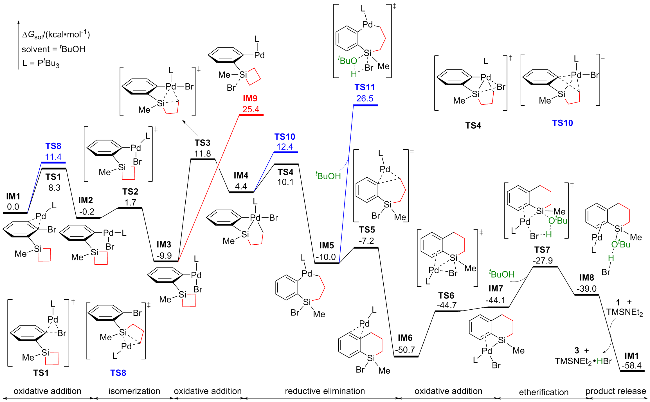
从IM3开始提出了两条反应路径: (1) Br/OtBu配体交换. 首先考虑tBuO-的生成, TMSNEt2与tBuOH经过四元环过渡态TS12生成TMSOtBu和HNEt2, 需克服40.2 kcal/mol的能垒. 随后, TMSOtBu解离形成tBuO-, 该过程吸热86.1 kcal/mol, 表明形成tBuO-非常困难, 因此tBuO-与Br-的配体交换不可能发生. 进一步研究表明, tBuOH与Br-的配体交换过渡态并不存在, 且能量升高. 接着, 我们考虑TMSNEt2协助的tBuOH/Br-配体交换, 其通过过渡态TS9或TS13形成Pd(II)-OtBu中间体, 此过程吸热26.6 kcal/mol, 且TS9或TS13的活化能比TS3高17.8或25.7 kcal/mol, 表明该路径在动力学和热力学上均不可行. 因此, IM3很难发生Br/OtBu配体交换且副产物5无法形成(详细的实验数据和图表在辅助材料中). (2) Pd—Br键与硅环发生分子内转金属化. 尽管进行了广泛的搜索, 仍未找到此过渡态. 于是, 我们提出了其他可行的反应路径. IM3通过过渡态TS3发生C—Si键的氧化加成, 生成五配位的Pd(IV)中间体IM4, 活化自由能为21.7 kcal/mol. IM3还可以异构化为五配位的硅中间体IM9, 但此过程需吸热35.3 kcal/mol.
IM4通过过渡态TS4和TS5依次发生Si—Br键和C—C键的还原消除, 生成Pd(0)复合物IM6. IM4转化为IM6的过程放热55.1 kcal/mol, 表明IM6的形成是不可逆的. IM4还可以通过过渡态TS10发生C—C键的还原消除, 但TS10的活化能比TS4高2.3 kcal/mol. 此外, 实验组提出Pd(II)物种IM5可通过过渡态TS11发生醚化, 但TS11的活化能比TS5高33.7 kcal/mol, 猜测是因为Pd中心没有参与此过程.
复合物IM6通过过渡态TS6发生Si—Br键的氧化加成, 形成Pd(II)物种IM7, 活化能为6.0 kcal/mol. 随后, IM7与tBuOH通过五元环过渡态TS7发生醚化, 得到底物3配位的Pd(0)中间体IM8. TS7的活化能为22.8 kcal/mol, 此过程涉及Pd—Si、Pd—Br、tBuO—H键的断裂以及H—Br和tBuO—Si键的生成, 并将Pd(II)还原为Pd(0). 我们也考虑了Pd-Si-tBuO-H四元环过渡态TS14, 但其活化能比TS7高8.5 kcal/mol. 最后, IM8通过配体交换释放目标产物3, 并再生IM1.
综上所述, 实验组提出的两条路径在能量上均不可行, 因此提出了一个新的催化循环: 底物1首先通过 C—Br键的氧化加成、C—Si键的氧化加成、Si−Br键的还原消除及C—C键的还原消除四个步骤, 经历Pd(0)/Pd(II)/Pd(IV)/Pd(0)过程, 接着通过Si—Br键的氧化加成及tBuOH的醚化两步, 经历Pd(0)/Pd(II)/Pd(0)过程得到产物3, 其中醚化过程是速率决速步骤, 活化自由能为22.8 kcal/mol.
1.3 Pd催化的环戊硅烷开环/交叉偶联反应机理
在明确环丁硅烷开环/交叉偶联反应的机理后, 我们将研究焦点转向环张力较小的环戊硅烷, 并以Scheme 1中的第二个反应为模板反应, 其Gibbs自由能如图2所示. 与环丁硅烷体系类似, 该反应的势能面以底物2配位的Pd(0)物种IM1A作为起点. IM1A通过过渡态TS1A进行C—Br键的氧化加成, 并经历Y型过渡态TS2A发生异构化生成Pd(II)物种IM3A, 该过程放热11.0 kcal/mol. 此外, 也考虑了C—Si键氧化加成路径, 但其对应过渡态TS7A的活化能比TS1A高10.7 kcal/mol. 同时, TS7A活化能比环丁硅烷C—Si键氧化加成过渡态TS8高5.8 kcal/mol, 这与环戊硅烷的环张力低于环丁硅烷的特性相符.
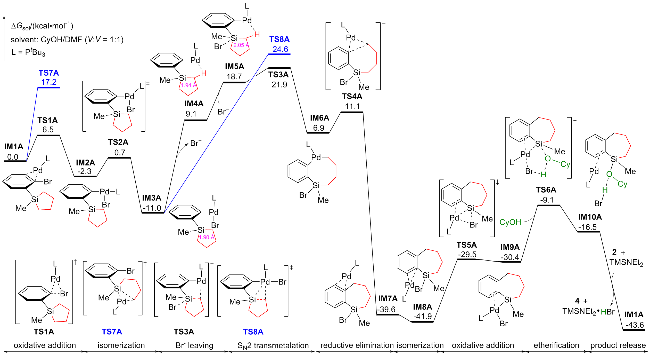
类似于环丁硅烷, IM3A可通过过渡态TS8A发生 C—Si键的氧化加成开环, 但需克服35.6 kcal/mol的能垒. 为寻求更可行的路径, 考察了实验组提出的Br/OtBu配体交换. 尽管TMSNEt2协助tBuOH/Br-配体交换的过渡态TS10A活化能比TS8A低4.8 kcal/mol, 但后续生成产物4的σ-键复分解过渡态TS11A能量高达57.7 kcal/mol. 同时可能生成副产物6或7的C—O键还原消除过渡态TS14A、β-H消除过渡态TS12A能量也在39.9 kcal/mol以上, 表明副产物6或7难以形成, 环戊硅烷可能通过其他模式开环.
通过计算发现IM3A可以离去Br-生成中间体IM4A. 通过对Pd—Br断裂的过程进行势能面扫描, 发现IM3A到IM4A是一个能量上升的过程, 且不存在过渡态. 随后, Br-结合Si中心形成五配位的硅中间体IM5A. 从IM3A到IM5A, C—Si键从0.190 nm延长至0.205 nm. IM5A通过过渡态TS3A轻松开环, 仅需活化能3.2 kcal/mol. TS3A为SN2类型转金属化, 其活化能比TS8A低2.7 kcal/mol, 表明环戊硅烷倾向于通过形成五配位硅中间体开环. 随后, IM6A经历C—C键的还原消除生成Pd(0)物种IM7A. 接着, IM7A发生Si—Br键的氧化加成及CyOH的醚化, 最终释放产物4并再生IM1A.
综上所述, 在环戊硅烷的开环/交叉偶联反应中, 底物2首先发生C—Br键的氧化加成、SN2类型转金属化以及C—C键的还原消除, 经历Pd(0)/Pd(II)/Pd(0)过程,随后发生Si—Br键的氧化加成及CyOH的醚化, 经历Pd(0)/Pd(II)/Pd(0)过程, 最终生成产物4. 关键的SN2类型转金属化为速率决速步, 其活化能为32.9 kcal/mol, 所以环戊硅烷体系的反应温度高于环丁硅烷体系.
1.4 溶剂效应
通过对TS3A和TS8A的结构分析发现, TS3A中存在显著的电荷分离, 呈现两性离子特征, 而TS8A则未表现出这种特性. DMF的引入增强了溶剂的极性, 有助于TS3A过渡态的形成. 为验证这一假设, 选择了几种代表性溶剂, 并计算了TS3A和TS8A的相对Gibbs自由能(表1). 结果显示, 从非极性溶剂到极性溶剂, TS3A的活化能随着溶剂极性的增加而降低, 而TS8A则相反. 且TS8A与TS3A能量差越来越大, 表明DMF的加入显著促进了环戊硅烷通过SN2类型转金属化开环.
表1 TS3A和TS8A在各种溶剂中的相对Gibbs自由能Table 1 Gibbs free energy of TS3A and TS8A in various solvents |
| Solvent | εrb/(F•m-1) | ΔG≠/(kcal•mol-1) | ∆∆G≠(TS8A-TS3A)/(kcal•mol-1) | |
|---|---|---|---|---|
| TS3A | TS8A | |||
| THF | 7.4 | 34.2 | 34.4 | 0.2 |
| tBuOH | 12.5 | 33.4 | 34.8 | 1.4 |
| CyOH | 17.0 | 33.3 | 35.3 | 2.0 |
| Acetone | 20.5 | 33.0 | 35.1 | 2.1 |
| DMF | 37.2 | 32.7 | 35.2 | 2.5 |
| CyOH/DMF (V∶V=1∶1) | 32.9 | 35.6 | 2.7 | |
2 结论
利用密度泛函理论计算, 系统研究了Pd催化下环丁硅烷和环戊硅烷在开环/交叉偶联反应中的机理, 并提出了一条全新的反应路径. 如Scheme 3所示, 反应首先由底物配位的Pd(0)物种对C—Br键进行氧化加成, 生成Pd(II)-Br物种B. 随后, 由于环丁硅烷与环戊硅烷之间的环张力差异, 其开环机理有所不同: 环丁硅烷通过C—Si键的氧化加成开环, 形成Pd(IV)中间体F, 进而通过Si—Br键的还原消除生成Pd(II)物种G; 而环戊硅烷则通过SN2类型转金属化直接开环, 生成Pd(II)物种G. 最后, 中间体G依次经历C—C键还原消除、Si—Br键氧化加成和醇的醚化生成目标产物. 计算结果表明, 极性溶剂(DMF)的加入能够促使环戊硅烷通过SN2类型转金属化开环, 从而有效实现交叉偶联反应. 我们期待本研究中提出的新机理信息能够为未来开发过渡金属催化的环戊硅烷开环反应提供重要参考.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 计算细节
本文的所有密度泛函理论计算均使用Gussian 16量子化学计算程序完成[29]. 理论计算所涉及的反应物、过渡态、中间体和产物均采用色散矫正的B3LYP-GD3(BJ)密度泛函方法[30-34]进行优化. 其中, Pd、Br原子采用SDD基组[35-36], 其他主族原子(C、H、Si、O、P、N)则采用6-31G(d)基组. 为确保所有的反应物、产物和中间体无虚频, 而过渡态仅有一个虚频[33], 所有几何结构均在相同计算水平下进行简谐频率振动计算, 并提供298.15 K和101 kPa条件下的热力学数据. 内禀反应坐标(IRC)以确保每个过渡态都与正确的中间体相连. 在气相优化几何结构的基础上, 本研究进一步在连续可极化溶剂化模型(SMD)[37]中, 结合M06[38]泛函计算单点能量及溶剂效应, 计算中Pd和Br原子使用SDD基组, 其他原子采用6-311++G(d,p)基组. 对于环丁硅烷体系, 采用tBuOH溶剂; 对于环戊硅烷体系, 采用CyOH/DMF (V∶V=1∶1)的混合溶剂. 在讨论TS3A和TS8A的能量差时, 还考虑了四氢呋喃(THF)、CyOH、丙酮和DMF四种溶剂. 对于CyOH (eps=15.0, epsinf=1.465)和DMF (eps=37.219, epsinf=1.428)的混合溶剂, 按1∶1比例自定义了该混合溶剂的eps=26.1, epsinf=2.0963. 本研究中的Gibbs自由能是将在B3LYP-GD3(BJ)/6- 31G(d)、SDD(Pd)理论水平上优化几何结构获得的自由能校正值加到SMD(tBuOH、DMF)/M06/6-311++G(d,p)、SDD(Pd)理论水平下计算的单点电子能量中.
辅助材料(Supporting Information) 额外的计算数据以及中间体和过渡态的笛卡尔坐标. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(Zhao, C.)