

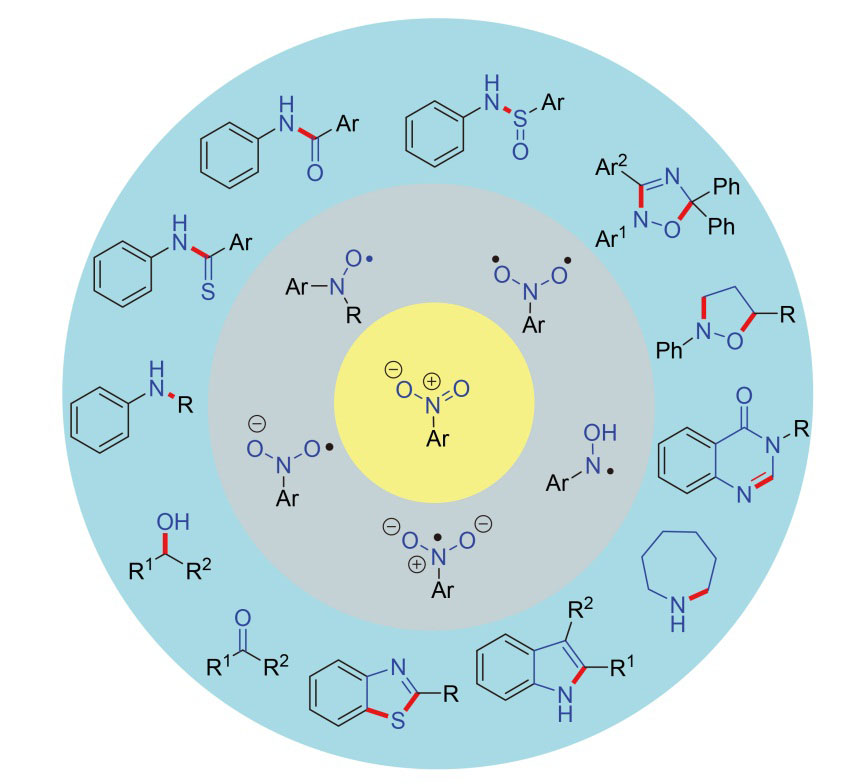
含氮化合物广泛存在于医药和天然产物中, 是药物化学中应用最广泛的一类化合物. 据统计, 目前上市的小分子药物中, 有90%以上的分子结构中都含有氮原子. 那些具有良好的抗炎、抗病毒和抗肿瘤等活性的分子也都包含氮原子[1]. 由于含氮化合物的广泛应用, 其合成也受到了广泛的关注. 如今, 在有机合成中大约每六个反应就有一个涉及到C—N键的形成, 传统的C—N键构建主要依赖于芳胺及其衍生物, 因为芳胺是C—N键形成的中心构件[2], 而获得芳胺最有效的方法之一是通过硝基芳烃的还原, 包括氢化、电子转移和氢化物转移等[3]. 与苯胺相比, 直接将相应的硝基芳烃构建C—N键可以节省一个或多个合成步骤. 此外, 胺、羟基和硫醇等官能团与硝基具有正交反应性, 因此它们可以在没有保护的情况下被耐受; 相反, 在使用苯胺的胺合成中, 必须保护这些官能团才能避免不希望的烷基化或芳基化等副反应的发生[4].
硝基芳烃化合物在有机化学的基础研究以及工业生产上的重要性是多方面的, 它既是化学工业的基本原料, 又在军事和化妆品方面具有重要作用, 并广泛用作染料、医药、农药和香料等精细化学品的生产. 与苯胺或亚硝基芳烃相比, 硝基芳烃作为底物的吸引力来自于它们在有机合成中的易得性. 硝基芳烃可通过Friedel-Crafts硝化反应[5]很容易地获得, 由于其廉价、易得和稳定的良好属性, 硝基芳烃已成为有机合成领域最理想、最重要的原材料之一. 由于硝基本身较为惰性的缘故, 硝基芳烃一般需要经过较为苛刻的还原条件将其先转化为其他化学中间体, 再对其结构进一步加工修饰才可获得目标产物. 例如, 早期研究者开发了利用(超)化学计量的膦[6]或一氧化碳[7]还原环合硝基芳烃以产生含氮化合物. 然而, 由于产生大量三氧化二磷废物和一氧化碳需要加压, 因此这类方法有明显的局限性. 随后许多过渡金属(Pd、Ru、Cu、Fe和Rh等)催化硝基芳烃构建胺类[8]、酰胺类[9]、磺胺类[10]以及吲哚等含氮杂环类[11]反应被实现. 此外, 有机硅烷(Organosilanes)作为新的潜在还原剂已经被人们所熟知. 研究发现, 有机硅烷[12]对于硝基是个较好的还原试剂, 可顺利地催化其还原串联反应[13]. 尽管如此, 上述这些方法在一定程度上仍受到过渡金属和昂贵配体以及特殊底物结构的限制, 有机硅烷的发现也属于初步阶段, 对很多含敏感官能团类型的硝基还原仍不耐受, 从而使底物范围有限. 随着绿色化学重要性日益显现, 发展简洁高效的原子经济型有机方法学必将成为今后化学研究的一大主题.
自由基因其活性高, 反应条件温和以及官能团兼容性好, 已被广泛应用于各类有机分子的合成和修饰[14]. 此外, 使用自由基中间体代替极性中间体时会出现许多其他合成的可能性, 这也使得自由基化学研究成为当代一个令人振奋的领域. 一般来说, 光催化的引入有助于自由基的产生, 而自由基的偶联反应扩大了可实现的转化, 这些方法比传统方法更具选择性和安全性[15]. 鉴于硝基芳烃原料的易得性和含氮化合物的重要性以及自由基化学的反应优势, 近年来自由基途径介导的硝基芳烃活化已经取得了较好的进展, 实现了多种多样的反应类型. 因此, 对该方面相关成果进行及时总结和综述具有重要意义.
目前, 关于硝基芳烃的催化活化反应已有部分综述报道, 反应类型包括以过渡金属催化[7b,16]、有机硅烷还原[12]以及可见光催化还原等为主[17], 而关于自由基介导的硝基芳烃活化反应的综述却未见报道. 本文便围绕并总结近十年来自由基介导的硝基芳烃活化反应, 并重点介绍了产生硝基氧自由基的方法, 然后基于单电子转移(SET)、能量转移(EnT)、氢原子转移(HAT)和电子供体-受体(EDA)复合物对这些自由基偶联机制进行描述与评估. 鉴于硝基芳烃还原为芳胺的类型较多, Verma等[18]关于光催化的硝基苯还原以构建苯胺也做了详细的总结, 此部分本文不作详细介绍, 只探讨个别最新的、经典的反应.
1 自由基介导的胺化反应
1.1 过渡金属催化的胺化反应
(杂)芳基胺是药物化学中一类重要的有机分子, 通常由苯胺合成, 使用苯胺-羰基还原胺化是最广泛的方法之一, 但不可避免的是使用化学计量的外部还原剂, 这些还原剂使得具有高度反应活性的基团化合物不可耐受, 如烯烃、羰基、羧酸、醛、酯和氨基等[19]. 苯胺大多又由硝基芳烃加氢合成获得, 因此由硝基芳烃直接合成(杂)芳胺具有良好的经济性和官能团相容性. 2015年, Baran等[4]报道了一篇由硝基芳烃和烯烃发生氢胺化反应合成叔烷基取代的芳胺的研究(Scheme 1). 该方法使用Fe(acac)3为催化剂, 苯基硅烷(PhSiH3)为氢供体, Zn为还原剂, 在温和反应条件下合成多种氢胺化产物, 产率中等至优异(96 examples; 24%~80% yields). 这种方法除了具有良好的普适性以外, 对许多未受保护的官能团都具有良好耐受性, 如羟基、氨基、酰胺基、酮羰基、三氟甲磺酸基以及硼酸基等. 此外, 该方案可以很方便地合成一些药物化学目标分子, 例如硝基吲唑两步法制备糖皮质激素受体调节剂中间体(52% yield); 硝基吡啶三步法合成HIV-1逆转录酶抑制剂中间体(43% yield); 硝基苯和酮缩合合成阿片样受体抑制剂中间体(37% yield). 尽管该方案在合成胺类化合物方面极其普遍, 适用于绝大多数底物以及具有良好的应用, 但仍有一定的缺陷. 例如硝基烷烃以及芳基乙烯类底物合成的烷胺产率极低; 2-硝基吡啶不耐受; 副产物较多, 包括硝基芳烃不完全还原导致的芳胺和N,O-烷基化副产物的生成, 为此, 通常需要过量的烯烃促使产率提升, 而整体的产物平均产率维持在55%左右. 初步机理实验结果表明, 虽然硝基芳烃在Fe(acac)3和PhSiH3体系下可以生成芳胺, 但直接使用芳胺作为中间体却无法获得目标产物, 因此排除芳胺是关键中间体的可能性. 最后证明烯烃氢胺化反应是通过硝基(杂)芳烃初始还原为相应的亚硝基(杂)芳烃, 然后与供体烯烃衍生的烷基自由基形成加合物.
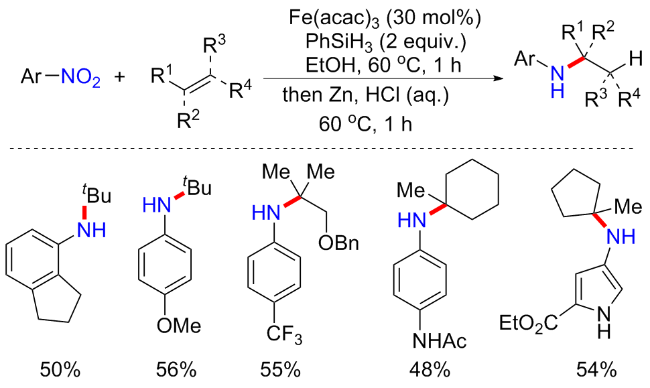
随后Thomas等[20]报道了一种在类似反应体系下(铁催化剂和硅烷)将硝基芳烃还原芳胺的反应. 该还原对硝基具有良好的化学选择性, 并对一系列功能底物(酮、酯、酰胺、腈、磺酰和芳基卤化物)具有良好的耐受性. 在此基础上, 进一步加入1,1-二取代烯烃, 通过烯烃的氢胺化反应构建了一系列的α-叔胺化合物. 研究证明该方案通过自由基途径进行, 亚硝基苯为关键中间体.
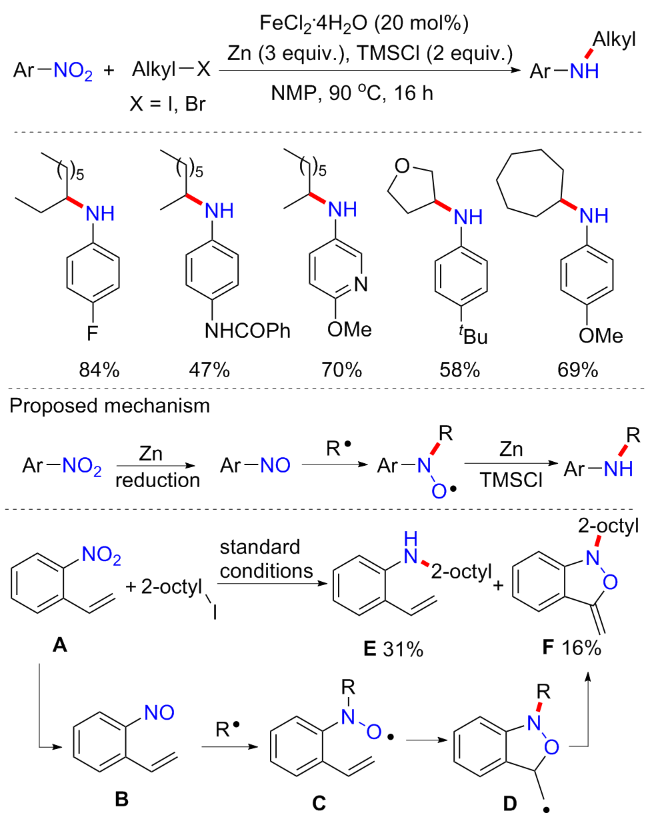
2016年, 胡喜乐等[21]开发了一种铁催化的硝基芳烃和卤代烷的还原偶联构建(杂)芳基胺方案(Scheme 2). 方案以N-甲基吡咯烷酮(NMP)为溶剂, 四水合氯化亚铁(FeCl2•4H2O)为催化剂(20 mol%), Zn (3 equiv.)为还原剂以及三甲基氯硅烷(TMSCl) (2 equiv.)为添加剂, 在90 ℃下反应16 h可获得最优产率. 在最优反应条件下具有非常好的底物普适性, 含有缺电子或供电子取代基的硝基芳烃都可合成相应的二级芳基胺; 二级或三级烷基卤化物也都具有良好的耐受性; 由于硝基与原子官能团的正交反应性, 羟基、氨基、酰氨基以及硼酸酯基等都具有良好的相容性. 有趣的是, 当烷基碘化物调整为1.5 equiv.并加入更多剂量的还原剂和添加剂(5 equiv. Zn和4 equiv. TMSCl), 可合成双烷基化的胺类化合物. 为了探究反应机理, 作者进行了一系列控制实验. 在不添加烷基卤化物情况下, 可产生少量苯胺产物, 但苯胺不与烷基卤化物反应; 当使用亚硝基苯时, 可以70%收率获得产物; 当使用2-乙烯基硝基苯作为底物时, 除获得预期的偶联产物外, 还获得异噁唑产物F; F的形成表明反应过程中产生了2-乙烯基亚硝基苯B. 可能机制为烷基自由基攻击B中的氮氧基团, 形成中间体C, 经过环合夺氢得到F. 因此, 上述实验支持亚硝基芳烃为可能的关键中间体. 此外, 2,2,6,6-四甲基-1-哌啶氧基(TEMPO)的使用, 观察到胺化产率显著降低; 当使用环丙基甲基溴作为自由基钟底物时, 获得了常规产物和开环产物. 最后, 受Baran[4]关于烷基自由基可以添加到硝基芳烃还原形成的硝基芳烃的提议的启发, 推测可能机制为: 铁(II)催化剂被锌还原成Fe(I), 然后激活烷基卤化物产生烷基自由基并再生Fe(II)[22]; 同时, 硝基芳烃被锌还原形成亚硝基芳烃; 接着烷基自由基攻击亚硝基芳烃的氮原子, 形成C—N键, 得到胺产物.
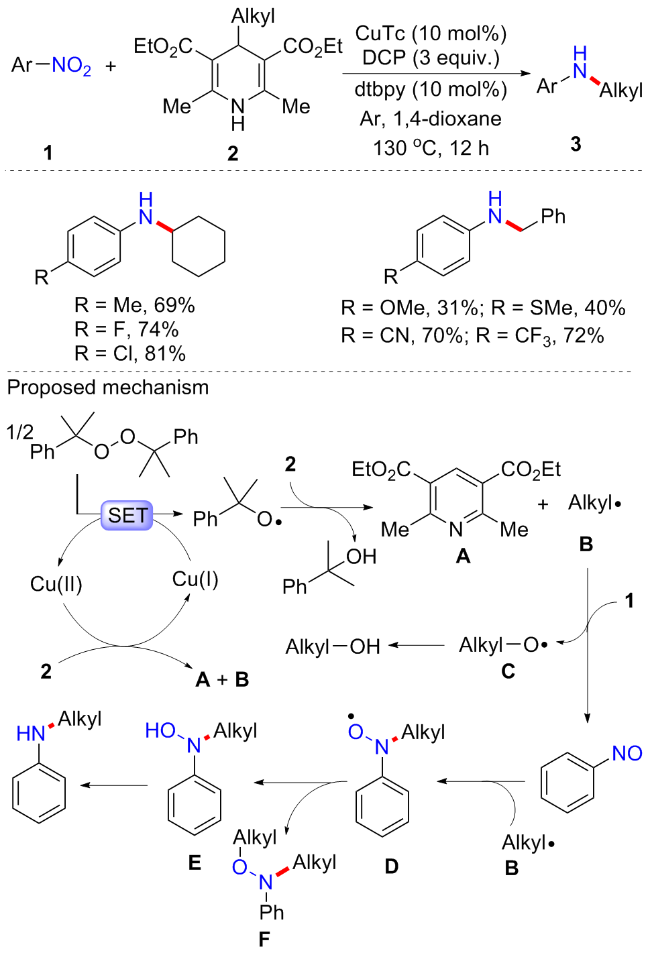
1,4-二氢吡啶可广泛用作还原胺化反应的还原剂, 由醛一步合成[23]. 考虑到4-烷基-1,4-二氢吡啶是一类(汉斯酯, Hantzsch ester)在氧化条件下或光催化下具有吸引力的烷基自由基前体, 并且与醛的还原胺化反应相比, 4-烷基-1,4-二氢吡啶的烷基化允许扩展到二级和三级烷基. 因此, 李金恒等[24]在2022年报道了铜催化的硝基芳烃与4-烷基-1,4-二氢吡啶的还原交叉偶联方案, 制备了烷基苯胺(Scheme 3). 该方案使用4-烷基-1,4-二氢吡啶作为烷基自由基源和内部还原剂, 为构建具有良好官能团耐受性的烷基苯胺提供了一个良好的互补平台. 反应普适性良好, 含苄基或环己烷等烷基取代的1,4-二氢吡啶都能以良好的收率合成目标产物; 含吸电子和给电子取代基的硝基芳烃也可耐受, 并且含醛基、酯、羰基以及氰基等官能团的底物也可相容. 但含供电子基的芳胺产率较低, 可能是因为含供电子基的芳基氮自由基更稳定, 停留的时间更长, 从而形成的N,O-烷基化副产物增多, 而硝基烷烃或硝基苯乙烯类的底物不可耐受的原因则可能是因为形成的氮自由基极不稳定. 控制实验表明, 反应可能经历了自由基介导的还原偶联机制. 首先, 过氧化二异丙苯(DCP)在Cu(I)存在下通过单电子转移(SET)产生烷氧基自由基和配合物Cu(II); 4-烷基-1,4-二氢吡啶通过SET发生C—C裂解生成吡啶A和烷基自由基B; 然后, 硝基苯与烷基自由基B的单电子还原产生亚硝基苯和烷氧自由基C, 进一步通过SET产生相应的醇; 值得注意的是, 烷基自由基不仅作为烷基来源参与反应, 而且通过SET还原硝基苯生成亚硝基苯; 随后, 烷基自由基B进攻亚硝基苯得到自由基中间体D, 最后还原为目标产物. 此外, 部分烷基自由基还可与中间体D偶联, 得到副产物N,O-烷基化产物F, 这个和Baran[4]的研究一致.
1.2 过渡金属与可见光协同催化的胺化反应
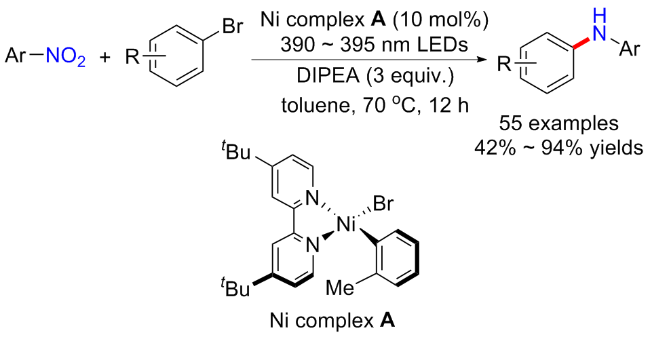
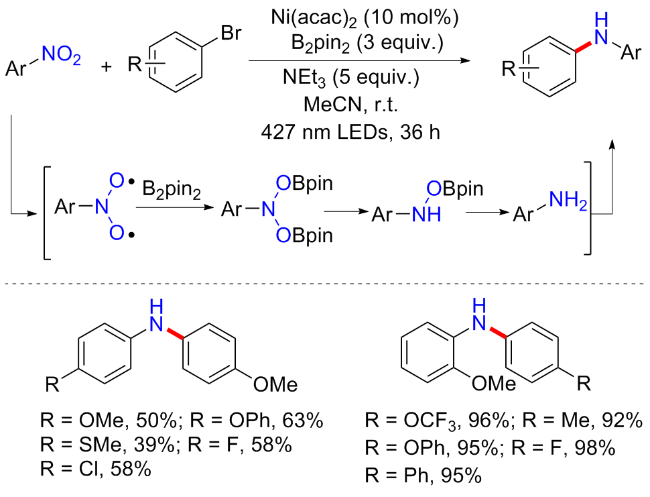
最近, 王雷锋等[26]实现了在无配体无光敏剂下, 可见光和镍促进的芳基卤化物和硝基芳烃还原构建二芳基(杂芳基)胺的方案(Scheme 5). 研究发现, 在B2pin2 (3 equiv.)和NEt3 (5 equiv.)存在下, 以硝基苯为模版底物, 乙腈为溶剂, 在室温下用蓝色LEDs (427 nm)照射36 h, 可以高产率地分离出所需的还原产物苯胺. 在此研究思路基础下, 作者通过添加10 mol% Ni(acac)2, 并以溴苯为底物, 高效地实现了硝基芳烃的级联还原与芳香族C—N交叉偶联反应. 反应普适性良好, 富电子或缺电子以及二取代的硝基芳烃和芳基卤化物均能耐受, 但总体产率中等, 并且硝基烷烃类底物仍旧不适用. 与以往研究不同的是, 控制实验表明苯胺为该反应的关键中间体, 并非亚硝基苯, 而偶氮苯、苯基羟胺等也不能以较好的收率得到目标产物. 整个反应过程分为两步, 第一步是硝基苯在B2pin2和NEt3作用下还原为苯胺; 第二步是可见光和镍催化的苯胺和溴苯的C—N交叉偶联形成二苯胺化合物. 紫外可见光谱证明了苯胺和金属镍可形成复合物, 在427 nm照射下产生激发态并发生分子内的单电子转移, 生成一价金属镍和苯胺自由基阳离子, 最后经氧化加成、自由基偶合以及还原消除得到二苯胺产物和二价金属镍, 完成催化循环.
硝基芳烃和烯烃、有机卤化物的新兴自由基还原胺化虽然已得到较好的开发, 但上述方案大多局限于仲胺的合成, 具有较大位阻的叔胺合成仍比较困难. 2022年, 谢劲等[27]开发了一种铁催化的硝基芳烃和和羧酸进行脱羧C—N偶联的高效策略, 可以35%~98%的产率合成广泛的芳香叔胺(Scheme 6a). 以苯乙酸和硝基苯作为模版底物, 筛选出最优反应条件: 在65 ℃蓝光照射下, 10 mol% FeI2, 1 mol% 4CzIPN, 10 mol%配体(L1)和还原剂(EtO)3SiH, 可以91%的分离收率得到所需的胺衍生物. 具有各种取代基的硝基芳烃和羧酸均能以良好的收率合成相应芳香叔胺, 但具有供电子取代基的底物较缺电子产率更高. 有趣的是, 当作者尝试两种不同取代的芳基乙酸和硝基芳烃进行三组分反应时, 成功地构建了不对称芳香叔胺(Scheme 6b). 但考虑到具有不同取代基的芳香族乙酸的脱羧速率和生成的自由基稳定性的差异, 会产生部分对称性芳香叔胺副产物, 因此这部分产率也相对较低(15 examples; 38%~59% yields). 控制实验表明, 亚硝基芳烃依旧是反应的关键中间体, 而蓝光照射和FeI2是反应的不可缺少条件. 根据实验, 可能的机制如Scheme 6所示: 硝基芳烃可被(TPP)Fe(II)催化剂与(EtO)3SiH还原形成Fe基配合物A; 通过可见光介导的羧酸阴离子单电子氧化或Fe-羧酸盐的光介导的配体-金属电荷转移(LMCT)过程产生的烷基自由基, 然后攻击A以形成 Fe(III)复合物B; 随后, 它可以与羧基阴离子进行阴离子交换以产生羟胺化合物D并再生铁复合物C; 羟胺化合物能够与(TPP)Fe(II)物种反应生成次级中间体F和(TPP)Fe(III) E; 在碘离子存在下, 所得的E和中间体F可以重新结合形成铁络合物G; 在蓝色LEDs照射下, G和烷基自由基发生SH2均裂过程, 生成叔胺并再生(TPP)Fe(II).
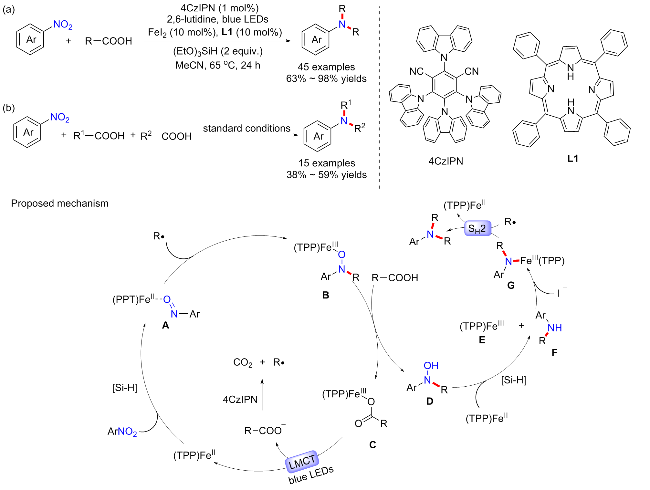
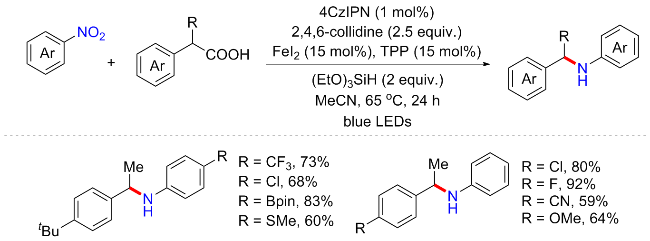
随后, 该课题组[28]继续开发了一种在相同催化条件下的协同光氧化还原和铁催化的硝基芳烃与羧酸构建仲胺方案(Scheme 7). 与此前不同的是, 以2-芳基丙酸替代芳基乙酸与硝基芳烃反应, 可避免生成的仲胺进一步与硝基芳烃偶联, 从而将产物停留在仲胺产物. 该方案同样可耐受广泛的官能团, 允许多种丰富的羧酸底物容易地合成二级胺(39 examples; up to 94% yield). 反应机制与Scheme 6类似. 烷基自由基可以通过羧基阴离子和激发的4CzIPN之间的单一电子转移或直接LMCT过程来脱羧产生, 并且LMCT途径是烷基自由基中间体产生的主要脱羧途径. 硝基芳烃在Fe(II)和(EtO)3SiH作用下生成亚硝基芳烃中间体, 随后与烷基自由基偶联并最终得到次级胺化产物.
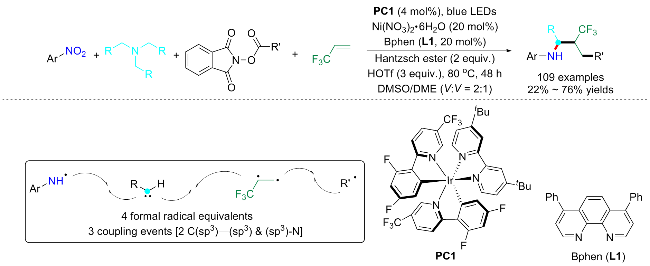
2023年, Cheung等[29]也以Hantzsch ester作为还原剂, 实现了一种金属镍协同光氧化还原促进的叔烷基胺、硝基芳烃和羧酸酯的三组分胺化反应(Scheme 8). 反应以[Ir(dF(CF3)ppy)2(dtbby)]PF6为光敏剂, Ni(dme)Br2为金属催化剂, 红菲咯啉(Bphen, bathophenanthroline)为配体, 使用Hantzsch ester (HE)作为还原剂, 三氟甲基磺酸(HOTf)作为添加剂, N-甲基吡咯烷(NMP)和1,4-二氧六环为溶剂, 使硝基芳烃选择性地与三乙胺(2.5 equiv.)和氧化还原活性酯(2 equiv.)反应, 提供所需的N-烷基苯胺产物. 值得注意的是, 双组分或二聚化副产物未被检测到, 显示了金属氧化还原催化在选择性多组分转化的能力. 最后作者对底物普适性进行考察, 尽管整体产率中等, 但普适性非常广(135 examples; 20%~84% yields). 经过大量对照实验, 提出机制可能如下: Ir光敏剂(IrIII)被蓝光激发以产生激发态IrIII*, 其可以通过HE单电子转移来传递高度还原的IrII物种; IrII还原硝基芳烃得到亚硝基芳烃A和N-芳基羟胺, 加入的HOTf添加剂可以提供外源质子, 以促进硝基芳烃的还原和脱氧反应; 亚硝基芳烃经过光催化还原形成N-羟基氨基自由基B; 此外, 叔烷基胺(2)被IrIII *氧化以及去质子化产生α-氨基烷基自由基C; 两者自由基偶联以提供中间体D, 并在镍或IrII催化下脱去一分子羟基得到E, E继续消除二烷基胺组分, 进而形成亲电N-芳基亚胺物种F; 氧化还原活性酯(3)(可通过羧酸与N-羟基邻苯二甲酰亚胺酯化获得)也被IrII还原并脱羧以产生烷基自由基; 最后, 化合物F与烷基自由基反应生成 N-芳基氨基自由基G, 最终经过IrII促进的单电子还原或通过氧化的HE (HE+•)的氢原子转移产生目标产物N-烷基苯胺产物.
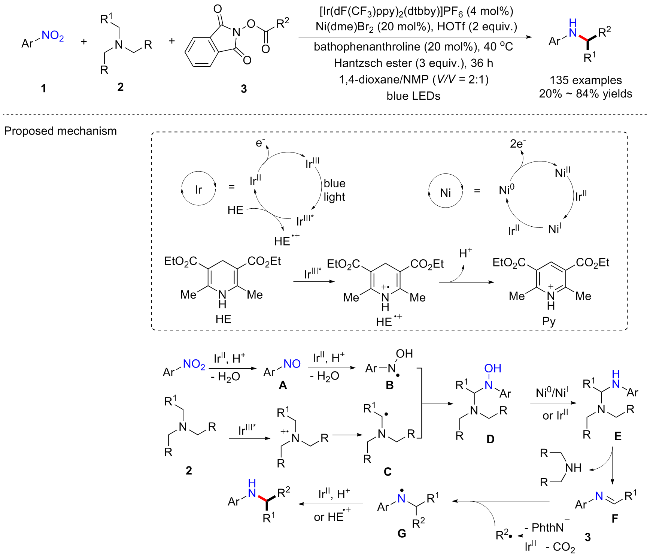
同年, Cheung课题组[30]继续以HE作为还原剂, 4CzIPN为光敏剂, NiBr2为金属催化剂, Bphen为配体, 在此反应条件下, 使硝基芳烃与三甲胺水溶液反应以提供N-甲酰基-N,N'-二芳基乙二胺化合物(Scheme 9). 尽管总体普适性良好, 许多复杂的底物都适用于该方案, 但产率均偏低(21%~68% yields), 而且一些简单的电子中性底物(2-硝基萘)、弱供电子底物(4-硝基甲苯)和吸电子硝基芳烃底物(1-氯-3-硝基苯、4-硝基苯甲酸甲酯、4-硝基苯甲腈)都不能合成目标产物. 推测这些硝基芳烃可能过度还原为其他非活性含氮物种, 如偶氮芳烃或者苯胺等, 从而抑制形成关键的亚硝基芳烃中间体, 而整体产率偏低也可能是因为硝基芳烃的过度还原导致. 此外, 目标产物N-甲酰基-N,N'-二芳基乙二胺化合物因其特殊的结构被证明具有良好的潜在应用. 如可用碱促进其去甲酰化提供对称的N,N'-二芳基乙二胺; 该对称产物还可与硫羰基二咪唑、双(三氯甲基)碳酸酯和苯甲醛进行环化, 得到相应的N,N'-二芳基咪唑烷硫酮、咪唑烷酮和2-(全氟苯基)咪唑烷. 具体反应机制与Scheme 8类似, 硝基芳烃提供目标产物的两个氮源, 三甲胺提供中间连接的两个碳源, 而溶剂中的水分子则提供氧原子形成甲酰胺基, 如Scheme 9所示.
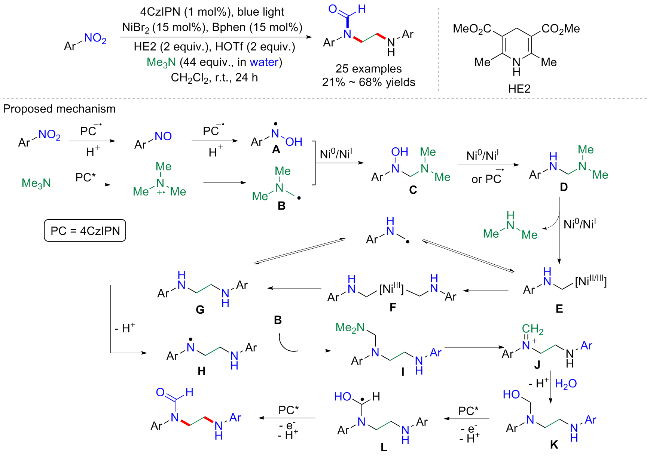
最近, Cheung和马军安等[31]联合开发了过渡金属协同光催化的三重偶联反应: 模块化合成复杂的N-三氟烷基苯胺(Scheme 10). 以3,3,3-三氟丙烯、硝基芳烃、叔胺和氧化还原活性酯为底物, 在上述相似的催化条件下(Ir为光敏剂, Ni为金属催化剂, Bphen为添加剂, HE为还原剂), 可以合成一系列具有三氟甲基基团和远端季碳基团的苯胺化合物. 尽管总体产率偏低, 反应时间也较长, 但普适性特别好(>100 examples). 另外, 使用该反应体系可以将脂质调节药物gemfibrozil和非甾体抗雄激素flutamide以及除草剂nitrofen转化为含有N-三氟烷基苯胺结构的衍生物, 甚至可以模块化合成脯氨酸羟化酶抑制剂的中间体, 用于治疗贫血. 这些例子证明了该方案在合成具有潜在生物活性的复杂分子方面具有良好的潜在应用. 其实4组分的反应研究在有机合成上还是比较少见的, 其机制探究也较为困难, 可能涉及多种分子之间的相互作用. 作者通过一系列实验包括自由基物种的检测、反应中间体和副产物的识别、氮素中间体的探索、α-氨基三氟烷基自由基的还原研究、Stern-Volmer猝灭分析等, 证实了光促进的自由基途经合成过程: 光催化剂Ir (PC1)在蓝光激发下激活, 还原硝基苯为亚硝基苯和N-芳基羟胺, 以及还原汉斯酯生成烷基自由基; 氧化态的铱物种氧化N,N-二甲基环己胺生成氮中心自由基阳离子, 然后去质子化生成N-环己基-N-甲基氨基甲基自由基; 最后, 烷基自由基与三氟丙烯反应生成三氟烷基自由基, 并与N-芳基氨基甲基自由基偶联生成N-三氟烷基苯胺产物.
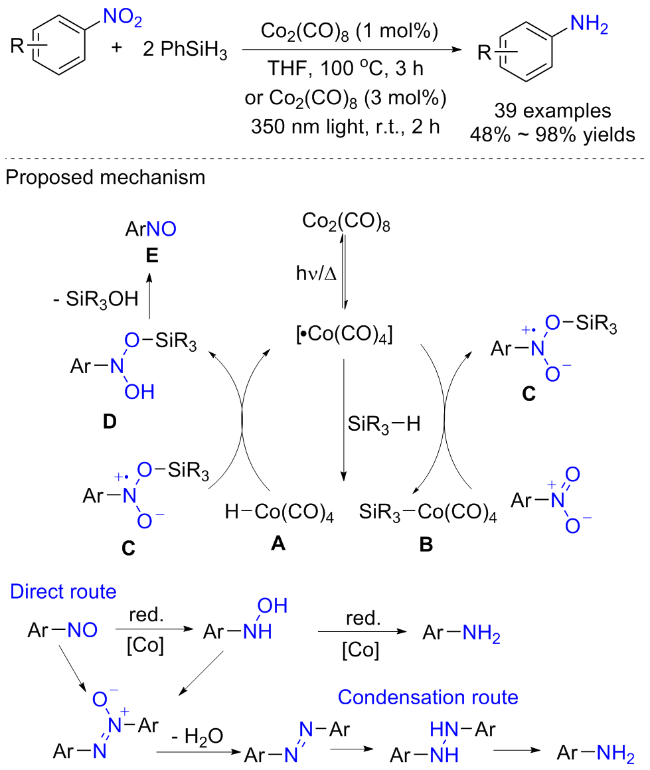
2023年, Bagh等[32]开发了一种将硝基芳烃还原成芳香胺的钴催化氢硅化方案(Scheme 11). 在热化学或光化学条件下, 商业可购的CO2(CO)8被证明是硝基芳烃还原的有效催化剂. 具有不同给电子和吸电子的硝基芳烃都是该方案的有效底物, 酯、氰基、酰胺和烯烃等还原基均可耐受, 体现了良好的化学选择性. 另外, 作者利用该方案顺利地合成了局部止痛药苯佐卡因、治疗麻风病的抗生素氨苯砜以及一些药物的中间体(用于治疗高血压的替米沙坦的起始原料、用于治疗疟疾的药物前体), 展现了该方案的实用性. 控制实验表明, 亚硝基苯是该方案的关键中间体, 生成的亚硝基可能经过两种途径还原为苯胺, 直接途径涉及亚硝基苯先还原为羟胺, 然后还原为芳香胺; 缩合路线涉及亚硝基苯和羟胺缩合生成氮氧基芳烃, 随后形成偶氮化合物, 最后通过肼中间体还原得到苯胺.
1.3 可见光促进的胺化反应
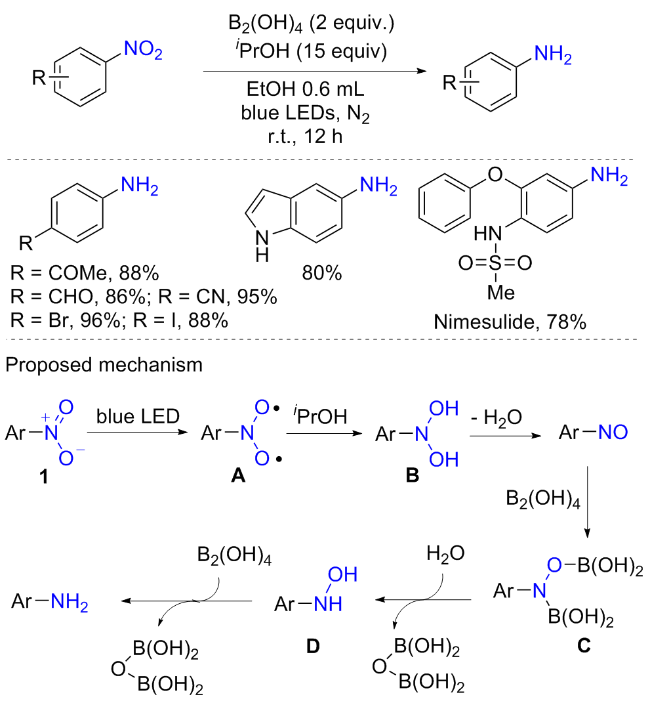
尽管上述过渡金属催化的硝基芳烃胺化包括部分协同可见光催化方案, 且具有较好的进展, 但无过渡金属催化的可见光促进的硝基芳烃胺化方案仍处于初步阶段. 2022年, 燕红等[33]报道了在无过渡金属催化条件下, 光诱导双氢原子转移对硝基芳烃进行化学和选择性位点还原生成芳胺的串联反应(Scheme 12). 该反应以异丙醇(iPrOH)为供氢剂, 四羟基二硼[B2(OH)4]为脱氧剂, 在室温光照条件下可实现超40多种芳胺的合成, 具有绿色、廉价和商业化等优点. 许多可还原的官能团, 如卤素、烯基、炔基、醛基、酮羰基、羧基和氰基都可耐受. 有趣的是, 同一分子中含有两个硝基的硝基芳烃具有良好的位点选择性. 例如, 氟化二硝基苯的邻硝基被选择性地还原, 对位硝基几乎没有被还原; 在二硝基取代的二芳基分子中, 缺电子芳基部分的硝基优先被还原; 结果表明, 底物的吸电能力越强, 反应速率越快, 产率也更高, 而供电子基的硝基芳烃则不太耐受. 通过改变溶剂和延长反应时间, 才获得个别中等收率的富电子芳香胺. 在控制实验和密度泛函(DFT)计算的基础上, 作者提出了如Scheme 12所示的反应机理. 首先, 光激发硝基化合物和异丙醇之间的双氢原子转移产生二羟胺B; 然后中间体B失去一个水分子转化为亚硝基芳烃中间体; 随后与二硼试剂的快速二硼化反应产生N,O-二硼中间体C(该中间体已得到核磁证实), 随后继续与水快速反应生成羟胺D; 最后经硼烷化脱氧得到最终产物. 此外, 可见光介导的硝基芳烃选择性还原为N-芳基羟胺也有报道过[34].
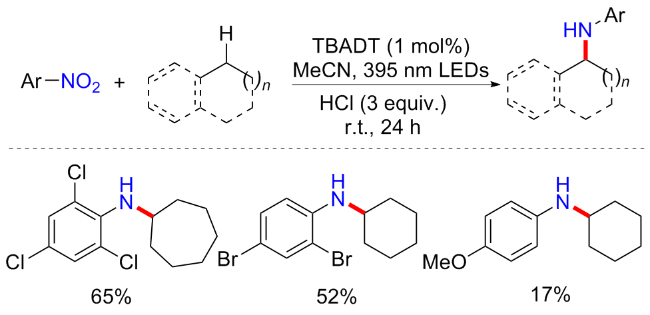
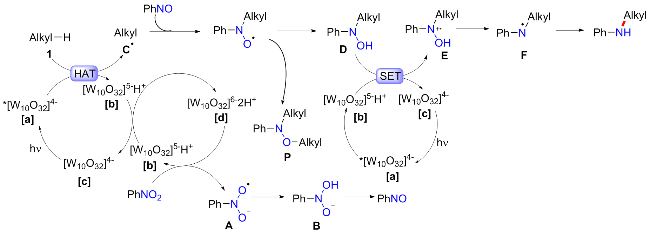
由于四丁基十钨酸铵(TBADT)作为商业上可获得的多金属氧酸盐, 可以用395 nm光照射从惰性烷烃中提取氢[35]. 王毅等[36]便开发了以TBADT为光敏剂, 可见光介导的硝基芳烃和未活化烷烃的交叉偶联方案(Scheme 13). 在最优反应条件下, 不同大小的环烷烃均能以良好的产率合成目标产物, 包括五元环、六元环、七元环、八元环以及金刚烷等; 含缺电子基的硝基芳烃产率较好, 而富电子取代的硝基芳烃产率则不尽人意; 同样, 杂环类的硝基芳烃虽然可以耐受, 但产率仅维持在11%~31%作用. 控制实验表明, 亚硝基苯为可能的中间体, 而苯基羟胺、偶氮苯只能提供微量的产物; EPR实验表明反应可能由自由基途径介导.
基于实验, 作者提出的可能机制如Scheme 14所示. 在光照条件下, *[W10O32]4- [a]的激发态与烷烃经过HAT反应得到烷基自由基, 并得到单一还原的十钨酸[W10O32]5-H+ [b]; [b]的歧化反应再生具有活性的光催化剂[W10O32]4- [c], 并同时形成双重还原的十碳钨酸[d], 十碳钨酸[d]对硝基苯进行单电子还原, 最初产生自由基阴离子A, 在溶剂中经过HAT反应生成B, 然后B失去一分子水生成亚硝基苯; 随后, 烷基自由基C与PhNO反应生成氧自由基, 经过HAT得到苯基烷基羟胺D; 最后, IV被[W10O32]5-还原为自由基阴离子E, 羟基的离去可获得氮自由基VI, 再经过HAT得到胺化产物. 烷烃的另一个被*[W10O32]4-氧化成自由基, 并与氧自由基偶联最终形成N,O-二烷基中间体P, 这也得到气相色谱-质谱(GC-MS)证实.
1.4 电化学促进的胺化反应
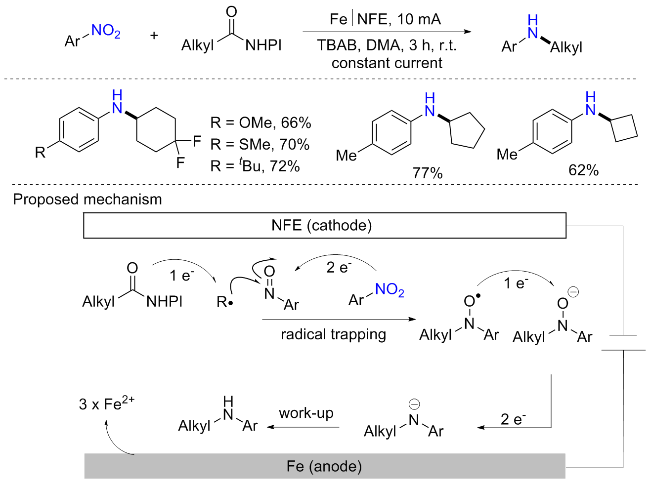
电合成化学概念的提出, 因其可使单电子还原且不依赖于昂贵的催化剂, 已经引起了学术和工业研究人员的广泛关注. 通过利用电化学的能量, 就有可能在阴极表面不断释放电子, 从而消除对化学计量金属还原剂的需求, 并避免其潜在的副作用[37]. 此外, 近年来以泡沫镍和泡沫铜等为代表的多孔泡沫型金属材料在电催化领域中显示出了非凡的应用潜力. 它们可以作为催化剂的支撑电极材料, 也可以通过修饰与改性直接用作催化电极. 因此, 电化学交叉亲电偶联方法为构建C(sp3)—N键提供了一个可持续和有效的手段. 2023年, 王毅等[38]开发了在无催化剂条件下羧酸酯和硝基苯之间的电还原偶联反应(Scheme 15). 反应以氧化还原活性酯和硝基芳烃为底物, 加入四丁基溴化铵(TBAB, 0.15 mol/L, 200 mg)、二甲基乙酰胺(DMA, 4 mL), 采用铁电极(52.5 mm×8 mm×2 mm)为牺牲阳极, 泡沫镍(NFE, 52.5 mm×8 mm×2 mm)为阴极, 在室温下, 以10 mA恒流搅拌和电解反应混合物3 h, 可以最优产率合成相应芳基胺化合物. 氧化还原活性酯由羧酸制备, 该方法能耐受1°、2°和3°的酸. 含富电子基和缺电子基硝基芳烃都可以相容, 以中等至高产率获得相应的产物. 利用该方案还可顺利合成抗人类巨细胞病毒药物, 以及含有异羟肟酸基团的脲酶抑制剂等, 体现了该方案的良好应用性. 实验证明亚硝基苯依旧是关键中间体, 在阴极优先还原硝基苯产生亚硝基苯, 并且烷基自由基的形成也发生在阴极, 这是由于在NFE阴极硝基苯的双电子还原导致的, 然后烷基自由基进攻PhNO产生氧自由基, 氧自由基经第二次单电子还原为氧阴离子, 然后在NFE阴极进行双电子还原得到芳基烷基氮负离子, 最后夺取溶剂中的氢合成最终胺化产物.
2 自由基介导的酰胺化及其类似物反应
2.1 过渡金属协同光催化的酰胺及磺胺化反应
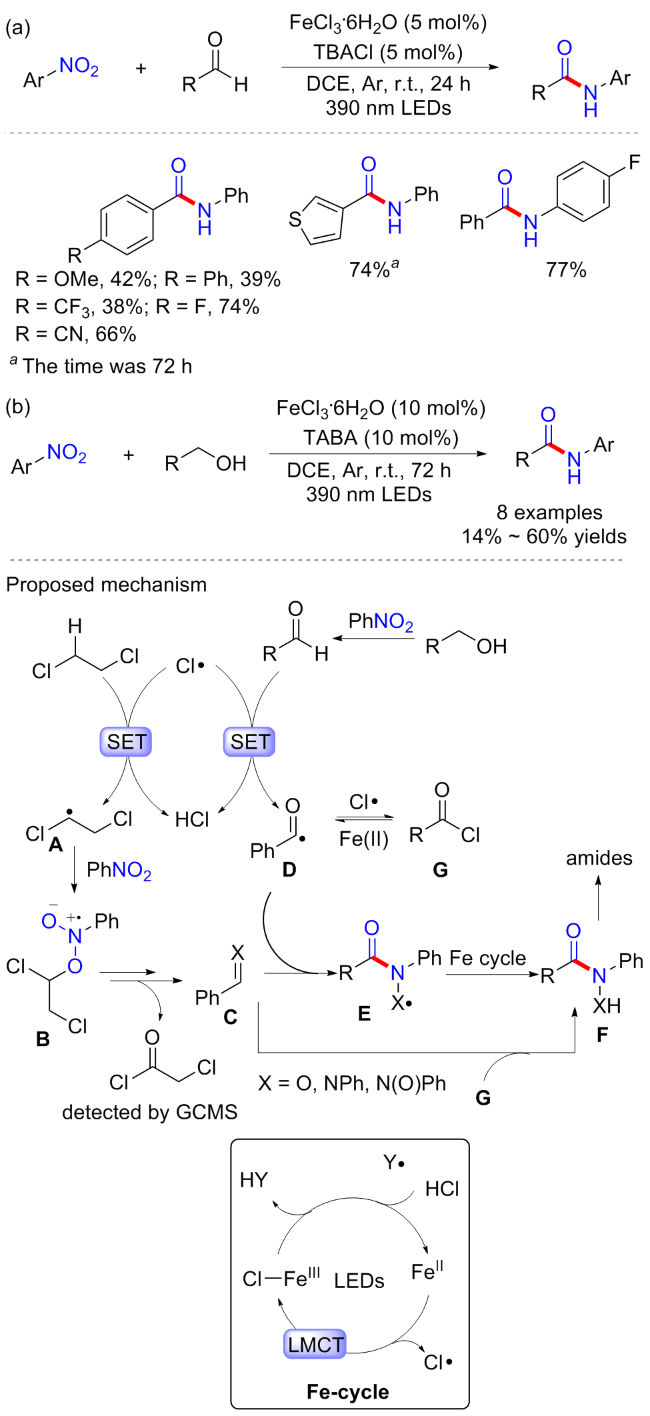
2022年, 曾荣等[39]报道了在室温氧化还原中性和pH中性条件下, 铁催化的硝基芳烃直接合成酰胺方案(Scheme 16). 该方案利用价廉易得的FeCl3•6H2O为催化剂, 四丁基氯化铵(TBACl)为添加剂, 协同390 nm的光照条件可实现芳香醛或脂肪醛与硝基芳烃的转化反应, 并且具有较宽的底物范围和较好的官能团耐受性. 有趣的是, 通过简单地改变反应条件, 将添加剂TBACl替换为TBAB, 用量从5 mol%增加到10 mol%, 可进一步促进醇与硝基苯的反应. 机理研究表明, 这些反应是通过氯自由基吸附产生光诱导的酰基自由基进行, 在转化过程中, 二氯乙烷溶剂作为O原子受体起着至关重要的作用. 当使用可能的中间苯甲酰氯代替苯甲醛时, 与硝基苯、亚硝基苯、偶氮苯和氧化偶氮苯的反应都成功地产生了酰胺产物. 结果表明, 所有的这些化合物都可能是潜在的中间体. 反应机制如下: 三价铁催化剂通过配体-金属电荷转移(LMCT)过程, 在光照下产生Cl自由基和Fe(II); 一方面, 氢原子在氯自由基和DCE之间转移, 生成了二氯乙基自由基A和HCl; 通过中间体B可以促进硝基苯还原为包括亚硝基苯、偶氮苯、氧化偶氮苯等可能的中间体C物种. 另一方面, 氯自由基还可以从苯甲醛中提取氢原子生成酰基自由基D, 它可以与C反应生成E, 也可以与氯自由基偶联生成酰氯G; 随后E在铁催化循环下将再生出三价铁和F; 最后, 在此还原条件下还原F得到酰胺产物. 当使用醇反应时, 乙醇最初会被硝基苯氧化生成醛是其可能的过程. 随后谢劲等[40]还报道了在FeI2、P(V)/P(III)和光氧化还原催化的三重协同催化下, 羧酸与硝基芳烃或硝基烷烃反应构建了结构多样的酰胺(86 examples, up to 97%).
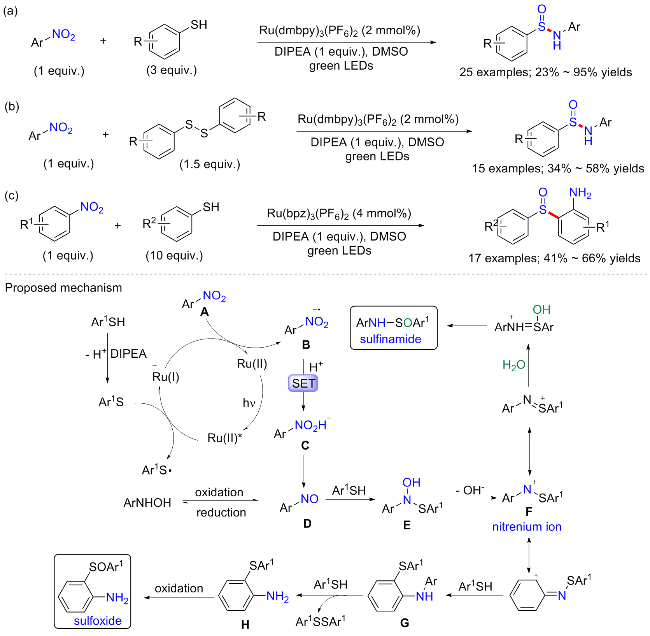
磺胺和亚砜类化合物是重要的有机硫化物之一, 也是天然产物、药物和除草剂中的典型结构单元, 在有机合成中发挥着关键作用, 被广泛用作合成中间体、配体和催化剂等[41]. 2022年, 曹剑瑜等[42]报道了一种以硫醇为硫源, 硝基芳烃为氮源的光催化合成N-芳基亚磺胺类化合物方案. 反应最优条件为Ru(dmbpy)3(PF6)为金属催化剂或光敏剂, N,N-2-异丙基乙胺(DIEPA)为碱, 3W绿色LEDs为光源, 在二甲基亚砜(DMSO)中反应10 h, 可以87%收率合成模板产物(Scheme 17a). 大量具有不同取代基的芳烃硫酚和硝基化合物都可得到相应的亚磺胺类化合物, 产率中等至优异, 但底物大多限于邻位有取代基的底物, 没有邻位取代基的苯硫酚产率较低; 而关于硝基芳烃, 那些具有给电子取代基的产物产率明显比吸电子取代基底物更高, 这是可能是因为缺电子的硝基芳烃很容易直接还原成苯胺, 而不需要在硝基芳烃阶段停留足够长的时间与硫醇反应. 有趣的是, 研究发现可以用二芳基二硫化物代替硫酚, 并以中等产率得到相应的亚磺酰胺产物(15 examples; 34%~58% yields) (Scheme 17b); 而随着反应体系中硫醇含量(3→10 equiv.)的增加, 反应途径的转变可以导致亚砜的形成, 从而为二芳基亚砜的合成提供了一种新的方法(17 examples; 41%~66% yields) (Scheme 16c). 控制实验表明, 亚硝基苯是该反应的关键中间体; 通过DMSO和18O标记的水作为溶剂, 证明了亚磺酰胺中的氧原子来自溶剂中的水. 具体反应机制可能如下: 苯硫酚在碱作用下脱氢成为质子供体, 并在钌催化剂下对硝基芳烃A进行双电子、双质子还原, 得到亚硝基芳烃D; D与N-芳基羟胺之间的氧化还原存在平衡, N-芳基羟胺或亚硝基芳烃与硫酚反应生成中间体E; E中N—O键裂解导致氮阳离子F生成, 它通过水的亲核进攻合成亚磺酰胺. 当硫酚浓度进一步增加时, 反应通过另一条途径生成亚砜, 在这种情况下, 阳离子F继续被其余硫酚进攻, 得到中间体G和芳胺H, 最后氧化提供亚砜.
2.2 无过渡金属催化的酰胺及硫代酰胺化反应
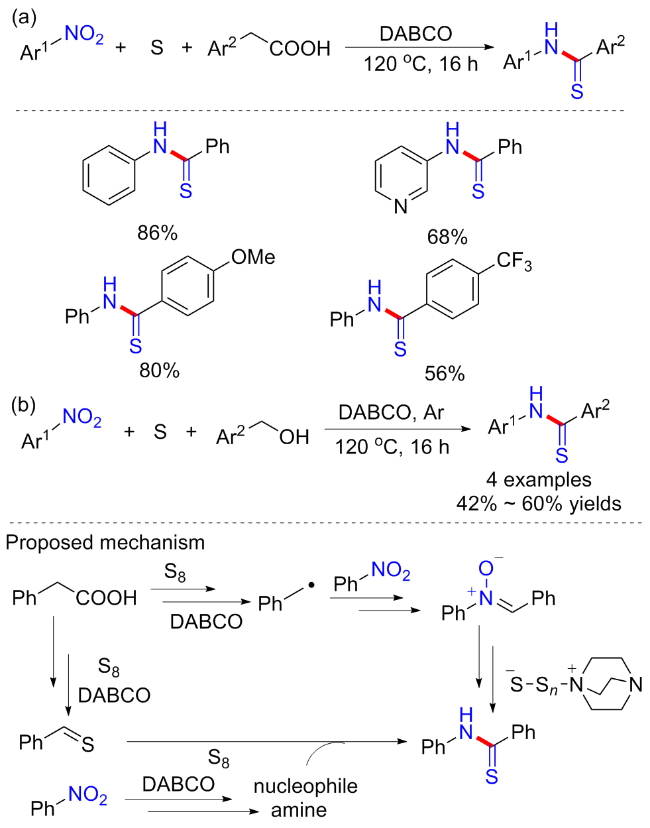
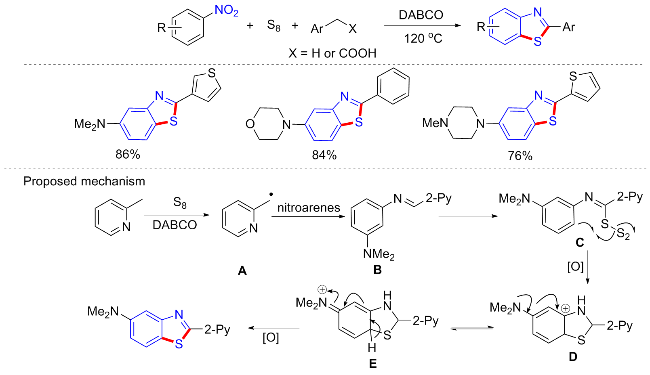
硫代酰胺结构广泛存在于生物活性化合物和药物分子中, 并且可以作为合成有价值的杂环或复杂结构的中间体[43]. 2019年, Phan等[44]报道了一种无过渡金属催化的硫代酰胺合成新方案. 反应不需要任何的催化剂或者配体, 甚至不需要额外的有机溶剂, 只需以三乙烯二胺(DABCO)为碱, 即可实现羧酸、硝基芳烃和元素硫的偶联(Scheme 18a). 具有供电子和缺电子的苯乙酸衍生物都可耐受, 但总体普适性一般, 且含供电子基的硝基芳烃不能合成相应产物. 研究发现, 苯乙醇在空气参与下, 也可在上述条件下与硝基芳烃反应合成硫代酰胺(Scheme 18b), 但普适性较差, 产率中等(4 examples; 42%~60% yields). 控制实验观察到一个由苄基硫醇自由基加成到二苯乙烯的产物, 在某种程度上证实了苄基自由基在反应中的相关性. 作者猜测反应机制很可能是通过硝基烷基中间体进行的, 但也不能排除先有苯并硫醛再有氮基亲核物淬灭的机制.
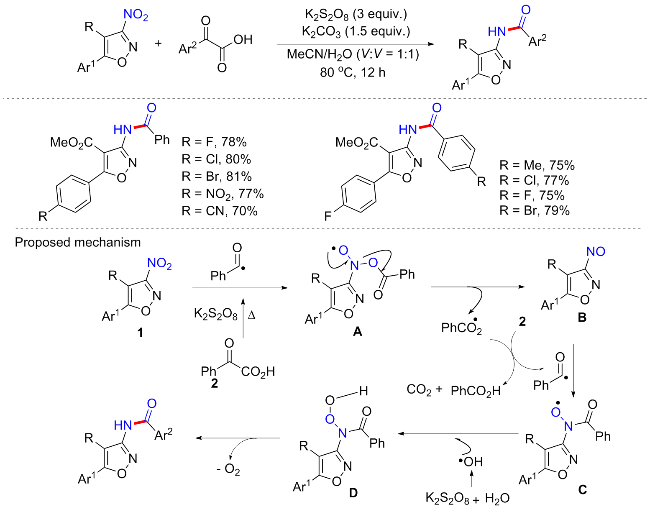
2020年, Batra等[45]研究了芳基α-酮羧酸与5-芳基-3-硝基异噁唑-4-羧酸酯在水相氧化条件下的脱羧/氧化酰胺化反应, 以提供N-芳基酰胺的方案(Scheme 19). 这种无金属和无还原剂且操作简单的方案具有良好的底物范围, 因异噁唑的C-4上存在一个吸电子基团, 使得C-3上的硝基对这些底物中的亲核取代反应高度敏感[46], 所以该方案适用于大部分C-4上存在羧酸酯或者羧酸结构的3-硝基异噁唑, 但C-4上含有酰胺结构的底物不可耐受. 并且只适用于C-5位芳基含缺电子取代基的底物, 含供电子基底物不可耐受. 实验证明, 该反应是由硝基芳烃和乙二醛酸苯甲酰自由基反应生成的关键中间体苯甲酰氮氧自由基与水中的羟基自由基偶联生成酰胺的自由基途径. 具体的可能反应机制为: 芳基α-酮羧酸在K2S2O8以及加热作用下生成苯甲酰基自由基, 用苯甲酰基自由基处理3-硝基异噁唑, 将提供苯甲酰基氮氧自由基C(通过亚硝基芳烃B); 接着由羟基自由基淬灭苯甲酰氮氧自由基C将产生一个中间体D, 该中间体D将进行原位脱氧生成所述的酰胺.
3 自由基介导的杂环化反应
3.1 可见光促进的杂环化反应
N-杂环化合物是各种生物活性物质和药物的结构基序或骨架, 尽管已经开发了相当多的这些骨架分子的合成方法, 但仍渴望以更高效率的策略来实现不同含氮杂环的构建, 同时最大限度地实现原子经济性. 鉴于硝基芳烃的广泛可用性和易于制备, 使用硝基作为氮源构建N-杂环已经成为许多研究者的目标. 2019年, 郑柯等[47]实现了邻硝基芳烃在温和条件下的可见光诱导的分子内环化反应, 为获得药物相关的喹唑啉酮衍生物提供了一种有效和环境友善的方法(Scheme 20). 该方案以邻硝基苯甲酰胺为底物, 在室温紫色光(395 nm)照射下, 以1.1 equiv. PhSiH3为末端还原剂, 以催化剂量的苯基硫脲为催化剂, 在干燥的1,4-二氧六环中反应可以最优产率获得喹唑啉酮衍生物. 苯基硫脲和PhSiH3的使用对于实现高收率是至关重要的. 底物普适性良好, 除六元环异喹啉结构外, 含五元环的异吲哚也可耐受, 并具有良好的产率. 作者通过对照实验、瞬态荧光、紫外-可见光谱和DFT计算在内的机理研究, 表明通过分子内单电子转移形成活性双自由基中间体是催化循环的关键阶段. 环化过程具体可能如下: 首先, 邻硝基苯甲酰胺经1,7-HAT产生酸-硝基互变异构体中间体B, 接着硫脲通过氢键与中间体B相互作用, 形成分子络合物中间体C; 光活性中间体C经过分子内单电子转移(SET)形成三重相双自由基中间体D, 然后, 自由基对的重组保证了C—N键的形成并伴随着六元环中间体E的构建; 硫脲中的碱性N原子从E中提取H原子, 得到中间体 F; 最后, 通过PhSiH3还原得到喹唑啉酮产物.
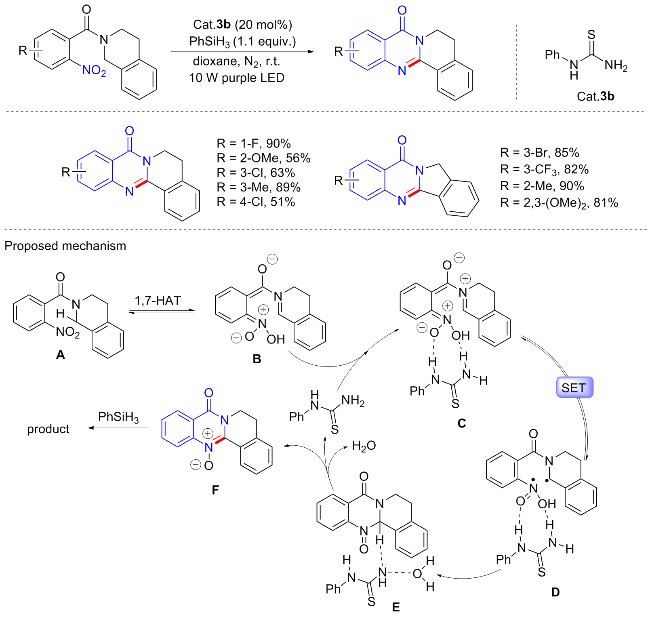
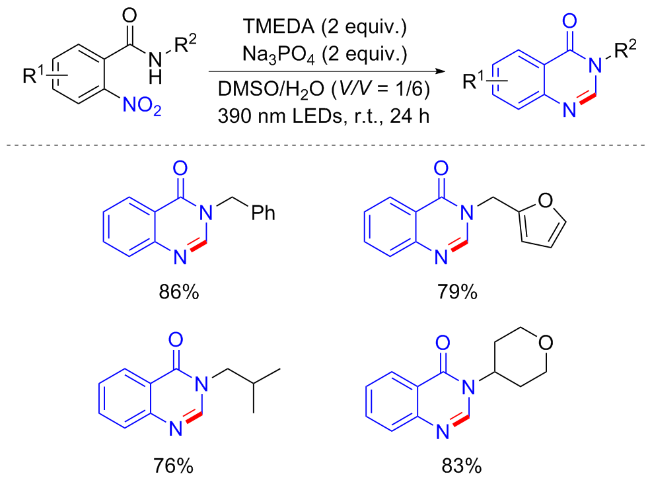
最近, 金灿等[48]实现了2-硝基苯甲酰胺衍生物合成喹唑啉酮的新方法(Scheme 21). 在光照催化条件下, 除了六元环异喹啉取代的2-硝基苯甲酰胺能自身环化合成喹唑啉酮外, N-苄基-2-硝基苯甲酰胺类似物也是良好的底物. 通过改变实验条件, 以四甲基乙二胺(TMEDA)和Na3PO4作为添加剂, 有利于喹唑啉酮骨架的合成. N-芳基取代衍生物包括苯、吡啶、萘和呋喃都以中等至良好的收率产生环状产物; N-烷基取代衍生物包括异丙基、环己基、正丁基等都可耐受. 有趣的是, 将底物替换为N-苄基-2-硝基胺类似物, 还可实现多种苯并咪唑衍生物的合成. 实验证明, 分子内的自由基偶联是该反应的可能机制.
2020年, 鲁桂等[49]报道了光催化的硝基芳烃和芳胺光氧化还原合成亚甲基硝酮中间体, 高反应活性的亚甲基硝酮进一步与烯烃环加成生成各种异噁唑烷衍生物(Scheme 22a). 这种三组分反应的特点是使用N,N-二甲基苯胺或N-芳基甘氨酸作为C1构件经[1+2+2]环加成, 在温和的条件下可以得到大量有用的异噁唑烷类化合物, 产率中等至良好. 当氘化的N,N-二甲基苯胺进行标准反应时, 以62%的收率获得CD2标记产物, 表明N,N-二甲基苯胺是异噁唑烷产品的C1构件. 具体反应机制可能为: N,N-二甲基苯胺和硝基苯在光催化下经SET产生硝基自由基A和氮自由基B, 二者通过氢原子转移生成D, 而A经过还原得到亚硝基苯并进一步产生苯基羟胺; 随后苯基羟胺进攻D生成关键中间体亚甲基硝酮.
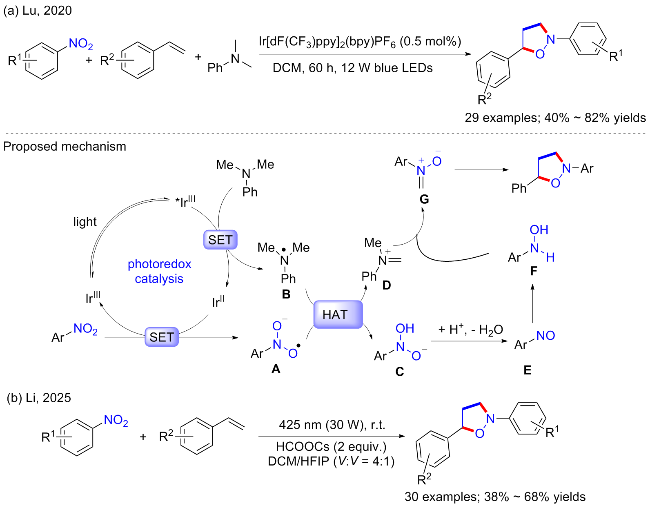
最近, 李加洪等[50]也报道了可见光催化的硝基芳烃和烯烃之间的[1+2+2]环加成反应, 在温和的条件下合成多种异噁唑烷衍生物(Scheme 22b). 与上述不同的是, 该方法不需要光催化剂或过渡金属催化剂. 机理研究表明, 在光照下烯烃与三重态硝基芳烃相互作用形成硝酮中间体, 作为C1源, 随后与烯进行[3+2]环加成反应得到所需产物.
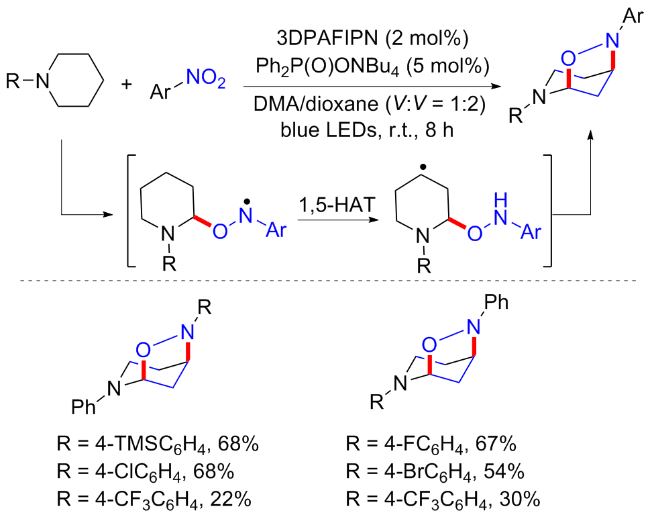
随后, 周其林等[51]利用N-苯基哌啶代替N,N-二甲基苯胺, 实现一个新的可见光诱导的α,γ-C(sp3)—H双官能化构建异噁唑反应(Scheme 23). 这种氧化还原中性、原子经济的方案具有广泛的底物范围和良好的官能团相容性, 是构建含哌啶桥环分子的一种简洁、实用的方法. 研究发现, 有机光催化剂必不可少, 并且2,4,6-三(二苯氨基)-5-氟异酞腈(3DPAFIPN)的产率优于Ir光催化剂; 由于质子偶合电子转移反应(pCET)已被证明可以促进强N—H键的均质化, 从而形成氮自由基, 作者便研究了各种碱作为添加剂, 发现Ph2P(O)ONBu4可以进一步将产率提高到85%. 反应普适性良好, 但总体产率中等偏低, 且硝基烷烃类以及含供电子取代基的硝基芳烃和苯基哌啶底物不可耐受. 机理研究表明, 通过原位生成的氮自由基的1,5-氢原子转移反应, 可以高度区域选择性地激活哌啶的γ-C(sp3)—H键活化.
2023年, 李漫波课题组[52]实现了光诱导的硝基芳烃和胺的氧化还原串联反应, 提供了高效率的异噁唑烷衍生物合成方案. 相比以前的方法, 该反应使用更为基础且惰性的三乙胺代替N,N-二甲基苯胺作为反应底物, 无需其他添加剂或还原剂, 在365~375 nm光照条件下可实现与硝基芳烃的高效偶联(Scheme 24a). 筛选条件发现, 紫色光照相比蓝光以及绿光, 具有更好的催化作用. 该方案底物普适性良好, 但缺陷是不可耐受含供电子取代基的硝基芳烃底物, 并且所有的产物均为对映体混合物, 使得其将来在药物化学领域的应用可能有限. N-苯基羟胺与乙醛或巴豆醛的成功反应表明, 该方案中间体很可能为三乙胺水解产生的乙醛. 硝基芳烃和三乙胺的光诱导氧化还原串联反应比N-苯基羟胺和巴豆醛之间的直接反应具有更高的收率, 这可能是由于在反应条件下羟胺和醛的不稳定性和部分产物分解. 这种比较进一步证明了光诱导氧化还原串联反应在实际应用中是有价值的. 基于控制实验, 作者提出了一个涉及光诱导氧化还原的可能机制: 首先, 硝基芳烃在光照下与三乙胺经过HAT过程生成硝基自由基和烷基自由基, 二者进一步氧化还原反应产生亚胺阳离子; 由于亚胺阳离子不稳定, 迅速转化为乙醛和亚硝基芳烃; 最后, 由乙醛缩合合成的巴豆醛和芳基羟胺经历Michael加成和环化以产生最终产物5-羟基异噁唑烷.
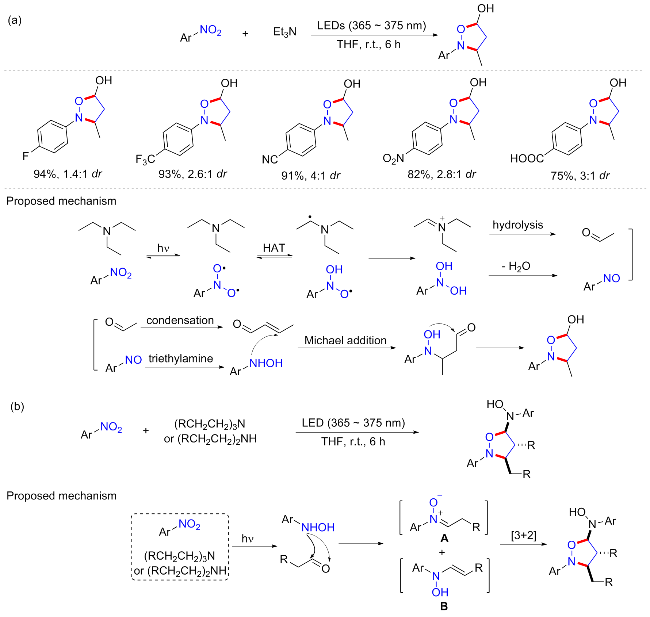
有趣的是, 进一步将其他烷基取代的三乙胺与硝基芳烃反应, 得到了不一样的产物. 作者发现其他烷基胺, 如三丙胺(二丙胺)、二丁胺、三丙胺(二乙胺)和三己胺(二己胺)都以5-羟基氨基异噁唑烷作为最终产物(Scheme 24b). 终产物的不同可能是在醛缩合阶段具有一定的化学选择性. 对于其中的三乙胺, 生成的醛的自身缩合是有利的, 提供与羟胺反应产生5-羟基异噁唑烷的α,β-不饱和醛; 相反, 对于其他烷胺, 空间阻力阻止了醛的缩合, 因此, 醛和羟胺之间的直接反应将分别生成A和B, A和B的[3+2]环加成反应得到5-羟基氨基异噁唑烷. 最近, 邓国军等[53]也报道了在相似条件下可见光诱导光氧化还原级联环化硝基芳烃与三乙胺, 构建异噁唑烷化合物.
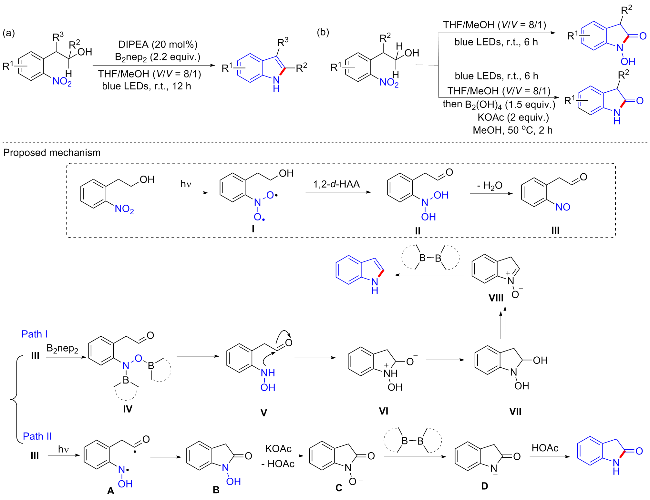
活化醇构建碳中心或氧中心自由基片段的光催化反应非常有助于醇类化合物转化为其他有价值的中间体[54]. 一般光催化HAT策略激活醇的富电子位α-C—H键通常通过氢键相互作用或羟基的去质子化来促进[55], 而激活醇的O—H键质子偶合电子转移反应(pCET)策略严重依赖于Brønsted碱与氧化剂的组合, 分别从O—H键中去除一个质子和一个电子. 基于此, 2022年, 燕红等[56]报道了一种光激发2-硝基芳基乙醇环化的策略, 用于构建包括吲哚、N-羟基吲哚酮和2-吲哚酮在内的N-杂环化合物(Scheme 25). 在不含过渡金属和光催化剂的条件下通过羟基烷基和硝基单元的分子内氧化还原促进C—N偶联. 该方案以DIEPA为催化剂, 双(新戊基乙二醇)二硼(B2nep2)为还原剂, 四氢呋喃(THF)/MeOH (V∶V=8∶1)为共溶剂, 在氮气下室温搅拌12 h, 以82%的分离收率合成吲哚(Scheme 25a). 有意思的是, 当反应不添加B2nep2时, 可以合成N-羟基吲哚酮, 而在弱碱性条件下, 通过原位生成的N-羟基吲哚酮进一步进行硼脱氧反应, 得到了一系列2-吲哚酮产物(Scheme 25b), 证明了反应在形成2-吲哚酮方面的可转换性. 此外, 利用该方案可以将氟他胺药物转化为其他吲哚类产物, 这为获取潜在的活性化合物或进行药物化学的结构-活性关系研究提供了一个有用的策略, 而不需要从头合成, 进一步证明了该方案的潜在应用性. 动力学同位素效应(KIE)实验表明, 碳氢键的均裂参与了速率控制步骤, 双夺氢反应(d-HAA)过程引发的环化反应可能是反应的可能机理. 首先, 基态硝基芳烃在适当的光照下, 被光激发成具有双自由基特征的三重态I, 然后活性双自由基参与相邻羟基的分子内双夺氢反应(d-HAA)产生亚硝基中间体III; 然后关键中间体III基于光照或硼催化剂可能按照两种不同催化途径进行, 具体如Scheme 25所示.
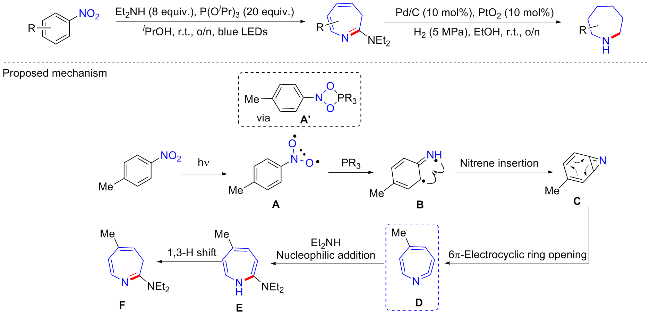
六元环的哌啶和五元环的吡咯烷是药物化学最常见的饱和氮杂环, 通过一些常见的含氮原料可以轻松制备[57]. 但是七元的氮杂环却非常少见, 合成方法也仅限于叠氮烷的环化或者功能化的哌啶贝克曼重排[58]. 最近, Leonori等[59]实现了以硝基为原料, 通过光化学脱芳环展开法制备氮杂环烷的方法(Scheme 26). 这个过程由蓝光介导, 以亚磷酸盐[P(OiPr)3]为催化剂, 在室温下用二乙胺处理硝基芳烃, 实现将六元苯骨架转化为七元环氮杂䓬(azepine). 接着在5 MPa H2下协同使用多相催化剂PtO2 (10 mol%)和Pd/C (10 mol%), 环保地产生环己亚胺(axepane). 这个还原过程相对于扩环过程在反应时间和收率方面都较差, 并且用于多取代氮杂䓬时, 还会导致非对映异构体的混合. 总体普适性良好, 含缺电子取代基的硝基芳烃较富电子基表现更好. 这样一个有趣且实用的反应机制可能为: 硝基在蓝光下产生硝基自由基A, 初步的计算表明, 双电子热激活过程可以得到四元环二噁唑磷酸乙酯A', 但它需要克服一个大的动力学障碍; 相比之下, 光激发和逐步自由基加成-重组是无障碍的. 所以, A'将经历热力学驱动的脱氧得到的亚硝基芳烃再进一步与磷试剂反应. 接着硝基的脱氧将导致关键的单线态氮化物B产生, 亚胺自由基共振形式的分子内环化将产生氮杂环化合物C, C经历6π-电环开环, 最终将全碳苯系统转化成七元环酮肟D. 由于D物质具有亲电特性, 使其能够与次级胺等亲核试剂进行简单的反应, 从而产生1H-氮杂䓬E. 然而, 由于E具有反芳香性质, 它将立即异构化成热力学稳定的3H-氮杂䓬F.
3.2 无过渡金属催化的杂环化反应
2021年, 我们课题组[62]实现了无过渡金属催化的2-氮杂烯丙基阴离子与硝基芳烃的[3+2]-环加成反应(Scheme 28). 该方案提供了一种绿色、操作简单的方法合成多种多样的2,5-二氢-1,2,4-噁二唑衍生物, 包括含各种不同电性的醛亚胺底物都可耐受, 含杂环以及多取代的硝基芳烃也是该反应的良好底物(>40 examples, 58%~95% yields). 反应不需要额外的还原剂、添加剂和催化剂, 仅需以叔丁醇锂作为碱, 在100 ℃下反应12 h即可实现高效的环加成反应. 对于以往关于硝基芳烃的研究, 需要过渡金属催化或需要可见光催化, 单纯的无过渡金属催化实现硝基官能团的活化实属罕见, 对此我们将注意力转移到了氮杂烯丙基这种特殊的底物. 根据前期我们课题组对氮杂烯丙基和醛亚胺的研究, 发现脱去质子后的2-氮杂烯丙基阴离子是一种超级电子给体(SED)[63], 或许是SED的性质活化了硝基芳烃. 基于相关控制实验以及DFT辅助计算, 该反应机制可能为: 醛亚胺的去质子化产生2-氮杂烯丙基阴离子, 从2-氮杂烯丙基阴离子到硝基芳烃的SET过程产生2-氮杂烯丙基自由基A和硝基阴离子B; 根据计算研究, 硝基芳烃自由基阴离子B不会与2-氮杂烯丙基自由基发生偶联, 相反, 在锂结合的硝基芳烃自由基阴离子和醛亚胺之间, HAT的势垒较低; 接着通过D消除氢氧化锂产生亚硝基芳烃和2-氮杂烯丙基自由基; 2-氮杂烯丙基自由基和亚硝基芳烃经过加成, 在F中形成C—N键; 随后中间体F经LiOtBu 脱质子化和异构化, 最终完成自由基的链传播以再生硝基芳烃自由基阴离子和噁二唑产物.
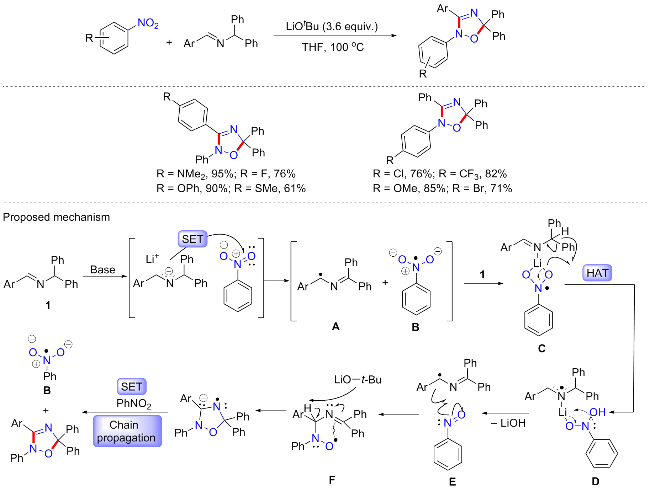
4 自由基介导的硝基氧转移反应
4.1 氧转移的羟基化反应
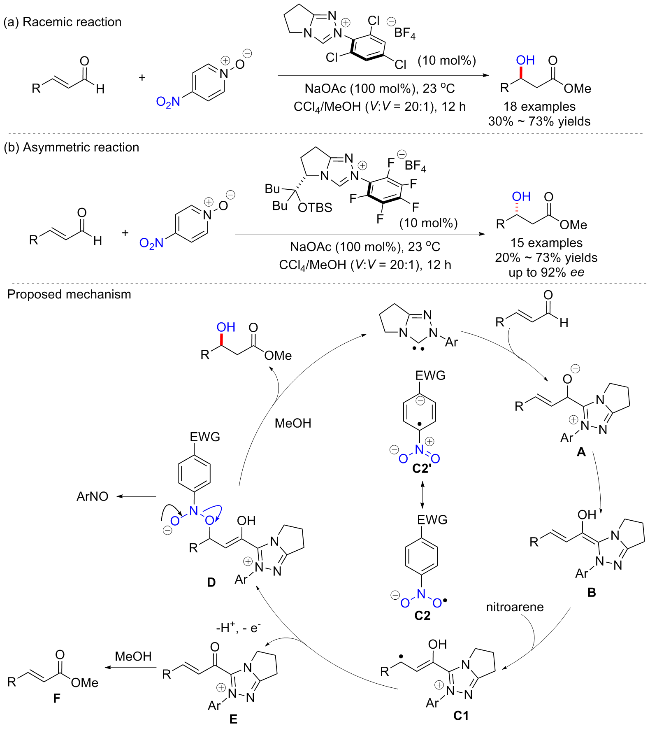
2014年, Rovis等[66]发现了以4-硝基吡啶N-氧化物为氧化剂, 加入卡宾催化剂, 在甲醇中加入肉桂醛, 可以得到相应的β-羟基酯(Scheme 29a). 作者最初以为氧转移来源于N-氧取代基, 在筛选了多种吡啶氧化物后, 发现只有含硝基的N-氧化物提供了产物. 关于硝基苯, 发现只有缺电子的硝基苯能够影响β-羟基化. 硝基苯衍生物的还原电位在反应活性中起着关键作用, Breslow中间体的氧化电位应该大于-0.49 V, 这也解释了为什么硝基苯($E_{\text{red}}^{\text{o}}$=-0.486 V)不能影响β-羟基化[67]. 反应的普适性较为广泛, 可以耐受芳基、杂芳基和烷基取代的肉桂醛. 但含有强富电子(OMe)或缺电子(NO2、F)的底物都导致较低的产率, 相反, 脂肪族底物可以提供较好的产率. 有趣的是, 通过改变手性催化剂, 当使用O-硅基保护的氮杂环卡宾(NHC)配体, 可以实现β-羟基酯的不对称合成, 并具有良好的对映体选择性(Scheme 29b). 作者提出, 反应应该首先通过形成Breslow中间体B进行, 随后经历硝基苯的单电子氧化提供Breslow中心的自由基阳离子C1和硝基苯衍生的自由基阴离子 C2; 然后这些自由基可以通过自由基偶合形成中间体D, 最后产生β-羟基酯产物, 并重生NHC配体. 随后, Chi等[68]以硝基苯磺酸氨基甲酸酯为氧化剂, 实现了N-杂环卡宾催化的肉桂醛β-羟基化反应. 催化反应允许将一个羟基引入到烯醛的β-碳中, 并高度对映选择性地合成具有重要用途的β-羟基酯.
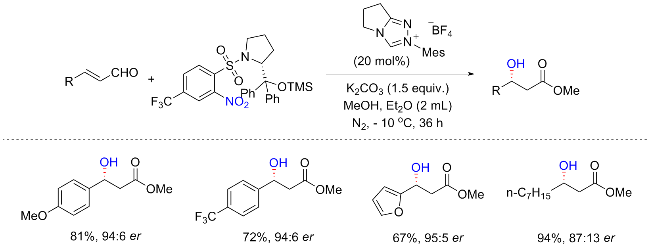
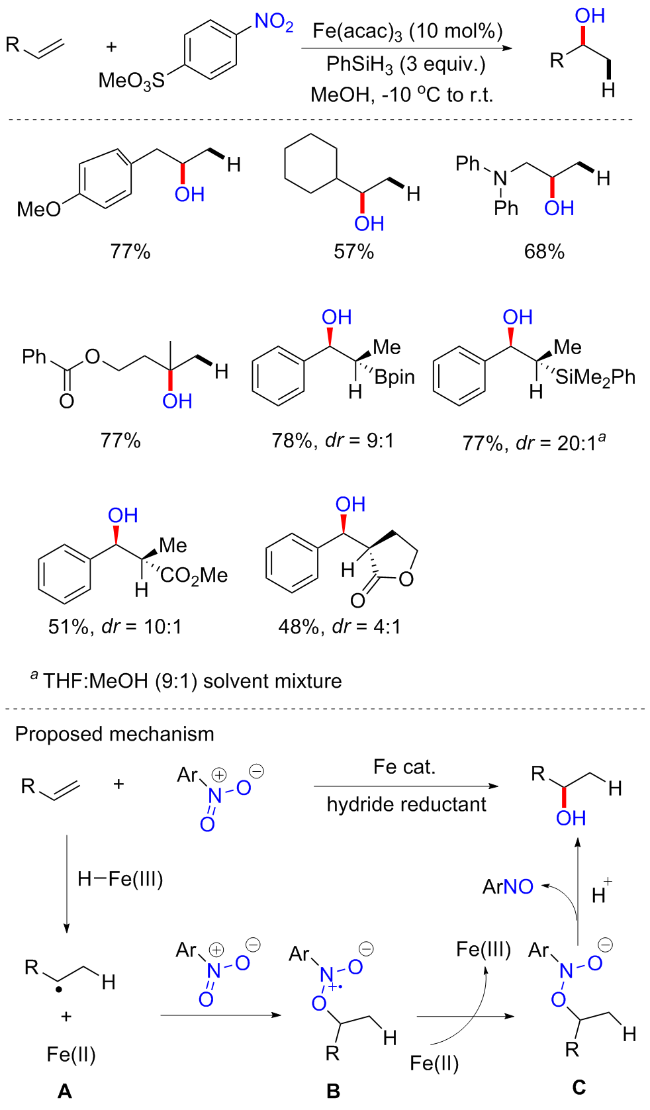
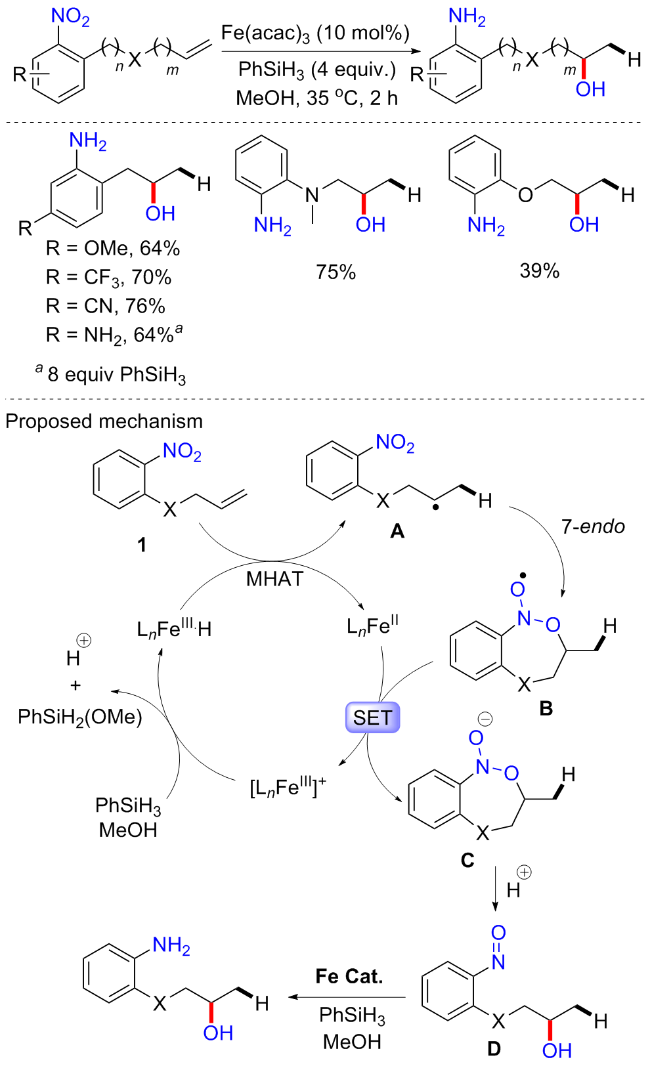
由于药物和天然产物中含有丰富的醇类官能团, 烯烃转化为醇类是一个非常重要的合成过程. 其实我们初步学习有机合成就知道, 烯烃氧化成醇的氧化剂常用冷稀KMnO4或OsO4, 显然, 它们的强氧化性以及毒性使得其应用越来越有限; 或者酸催化水的加入, 但它也容易导致副产物(醚的形成, 阳离子重排等)的形成. 2021年, Studer等[70]报道了以硝基芳烃为氧化试剂, 铁催化的非对映选择性的烯烃马氏(Markovnikov)水合反应(Scheme 31). 以烯烃为底物, Fe(acac)3为催化剂, 化学计量的PhSiH3为还原剂, 硝基芳烃为氧化剂, 在碱性条件下实现了多种醇的合成. 与上述研究一致, 在对位含有缺电子取代基的硝基芳烃对于烯烃的水合至关重要, 其中4-硝基苯磺酸甲酯提供了最好的结果. 芳香酚、胺、酰胺和硅烷等具有不同功能基团的脂肪族末端烯烃都可以耐受, 并提供中等至高产率的仲醇. 白桦脂醇、己酮可可碱以及一些天然产, 如β-香茅醇衍生的较复杂的烯烃可以在后期功能化, 以提供相应的高产量醇. 虽然这种反应可以得到很好的水合产物, 但是非对映选择性反应只能在刚性烯烃中实现, 有时需要非对映选择性的硝基芳烃以牺牲产率为代价来获得高的立体选择性. 机理可能为: 原位生成的 Fe(III)—H首先通过氢原子转移(HAT)与烯烃发生化学选择性反应, 得到相应的烷基自由基和Fe(II)物种; 然后中间物种B被Fe(II)物种还原, 形成所需的水合产物、亚硝基芳烃和Fe(III)物种.
随后, 该课题组[71]继续报道了一种利用硝基芳烃中的硝基官能团作为供氧体, 通过分子内氧原子转移到烯烃的自由基转化反应(Scheme 32). 反应在相似的催化条件下, 通过铁催化的金属氢原子转移到烯烃, 随后氧化导致烯烃水合. 与分子间自由基氧化反应相比, 分子内自由基氧化反应具有其他优点, 特别是能够显著改善非刚性烯烃水合反应的非对称选择性. 筛选发现, 较高剂量的PhSiH3 (4 equiv.)和温度的提升可以抑制分子间聚合副产物的产生. 该方案具有良好的底物普适性, 对于1,5-、1,6-甚至是1,8-氧转移都可以耐受, 而含有两个硝基取代的底物都会被还原为氨基. 对于分子间水合反应, 局限于缺电子硝基芳烃的催化, 但在分子内反应中, 富电子(OMe)取代的硝基芳烃底物也可耐受, 只是产率稍微有些降低. 实验表明, 反应很可能通过金属氢原子转移(MHAT)产生烷基自由基A, 在1,6-氧转移的情况下, 通过硝基官能团的NO双键上的7-内环化导致N-氧基自由基B的形成, 这种以杂原子为中心的自由基很可能进一步通过SET被初始MHAT步骤中产生的Fe(II)-复合物还原, 从而产生N-氧阴离子C; 然后, 质子化和开环将产生亚硝基芳烃D; 通过铁/硅烷催化体系进一步还原亚硝基芳烃最终产生水合产物.
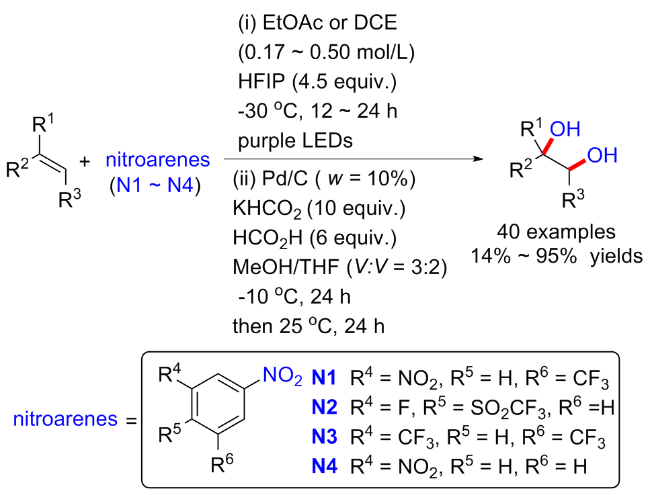
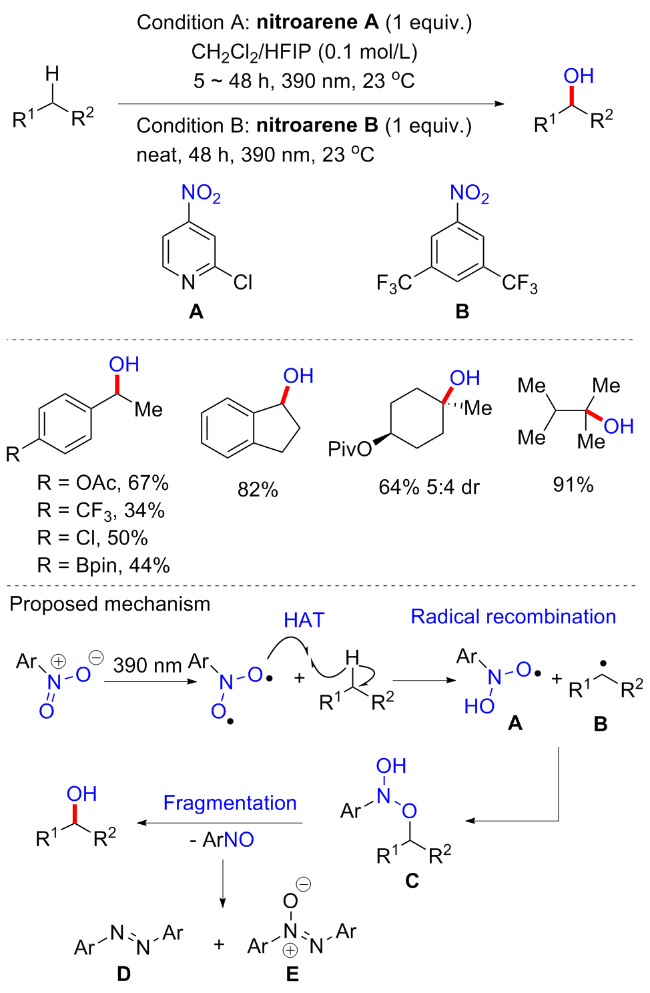
有趣的是, 硝基芳烃除了可以参与烯烃及其衍生物的氧转移水合反应以外, 还可实现苄位或未活化的C(sp3)—H键羟基化反应. 2023年, Parasram等[74]实现了在缺电子硝基芳烃存在下, 光激发硝基芳烃促进脂肪族体系的厌氧C—H羟基化反应(Scheme 34). 研究发现, 在缺电子硝基芳烃中, 2-氯-4-硝基吡啶和3,5-二(三氟甲基)硝基苯对于C—H键羟基化的效果最好; 六氟异丙醇(HFIP)作为添加剂, 对于抑制C—H羟基化产物的过氧化至关重要, 可能是氢键相互作用的结果. 富电子和中性底物表现良好, 可提供中等至良好的收率, 尽管具有吸电子基团的底物也适合于反应条件, 但产率较低. 值得一提的是, 在含有多个等效苄基位的底物中, 反应可选择性地产生单羟基化产物; 对于含有不对称苄位的底物, 苄位的氧化也是选择性的, 总体氧化反应性曲线表现: 二级(乙基苯)>一级(甲基苯)>三级(异丙基苯). 机理研究支持光激发硝基芳烃与碳氢化合物发生连续的氢原子转移和自由基重组反应, 产生羟胺醚中间体, 该中间体的自发裂解导致了关键的氧原子转移产物和亚硝基芳烃副产物的生成.
4.2 氧转移的酰基化反应
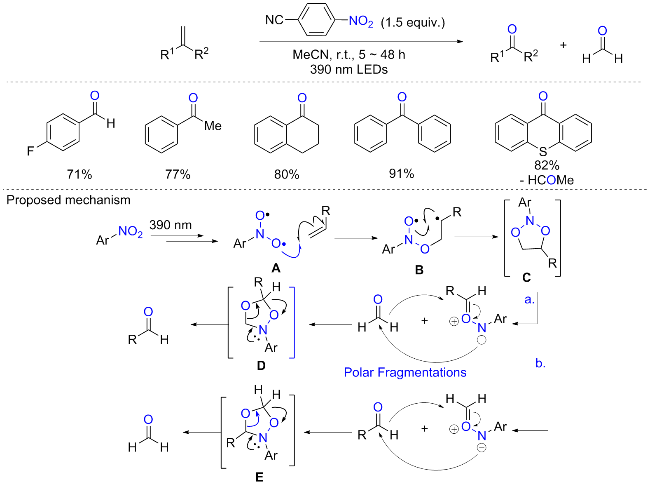
烯烃的氧化断裂合成羰基是有机合作中常见的方法, 常见的氧化剂包括臭氧、高锰酸钾、高碘酸钠等. 但是臭氧具有爆炸性以及难以控制化学计量, 而高锰酸钾的强氧化性使得化合物中的其他官能团易受影响, 并会产生了大量的有毒废物. 这些方法的局限性促使研究者开发更简便、环保的方法合成多样性的羰基化合物. 2022年, Parasram等[75]报道了利用硝基芳烃作为氧转移试剂, 在可见光下进行烯烃厌氧裂解制备羰基化合物的方案(Scheme 35). 以4-氰基硝基苯为最优氧化剂, 在390 nm紫光照射下可以实现烯烃的氧化裂解. 底物扩展表明, 含有给电子取代基和吸电子取代基的苯乙烯在该反应条件下表现良好, 相应的醛或酮产率较高. 多种具有敏感官能团(如游离羟基、羧基和炔基等)的烯烃耐受性良好, 经过厌氧裂解可生成具有高效率和区域选择性的羰基衍生物. 机理研究表明, 这种转化是通过光直接激发硝基芳烃, 然后与烯烃发生非立体定向的自由基环加成导致中间体C生成; 二噁唑烷C可以通过自由基途径或极性途径进行分裂, 通过自由基通路裂解的产物并未被实检测到, 而极性通路可以分为两种途径, 在任何一种情况下, 经过第一次开环后形成的羰基亚胺中间体和醛进一步经历1,3-偶极环加成反应产生1,4,2-二噁唑烷中间体(D和E), 随后开环生成所需的羰基产物. 自由基钟实验和原位核磁共振光谱分析证实了关键自由基种类和芳基二噁唑烷中间体的存在. 其实早在1956年, Ayer的开创性工作[76]便证明了用紫外光直接照射硝基芳烃可以导致一个更高的能量激发态, 从而触发与烯烃的环加成.
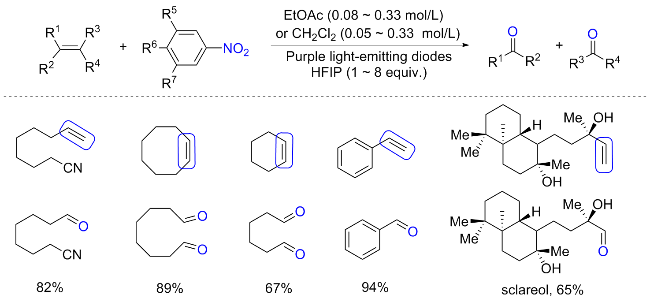
有趣的是, 在Leonori和Parasram的烯烃氧化裂解研究的基础上, Sarpong等[78]设想也使用光激发硝基芳烃, 从而实现肟的裂解, 这种裂解可以通过类似的机制进行. 对于环己酮衍生的酮肟与光激发的硝基芳烃反应也能生成自由基环加成中间体, 随后裂解产生酮羰基. 尽管确实实现了以硝基芳烃为氧化剂, 光催化的酮肟裂解合成相应的产物酮, 但是氘代实验表明, 产物酮的氧并非完全来自硝基官能团, 而是部分来自溶剂水或通过与水的快速交换.
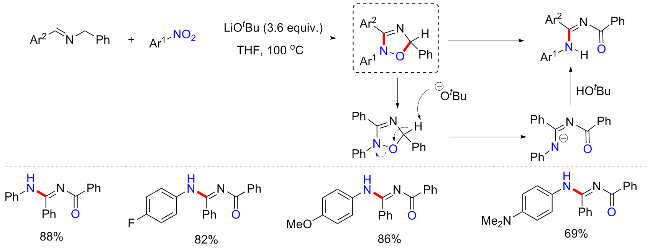
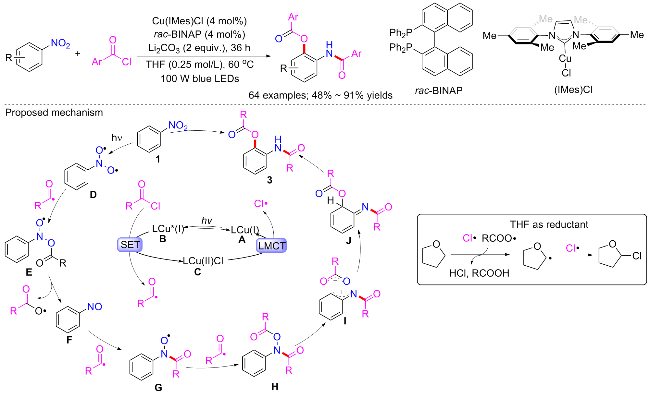
2024年, Ngai等[79]利用铜催化剂实现了硝基自由基和酰基自由基的偶合构建有价值的2-氨基苯酚衍生物(Scheme 38). 反应以硝基芳烃和芳基甲酰氯为底物, 以Cu(IMes)Cl (4 mol%)为催化剂, rac-BINAP (4 mol%)为配体, Li2CO3 (2 equiv.)为碱, 在100 W蓝光照射36 h, 获得了所需的2-氨基苯酚衍生物. 含富电子、电子中性和缺电子的硝基芳烃和芳基酰氯都可以提供期望的产物, 并提供中等至良好的收率; 脂肪族酰氯包括叔丁基酰氯和金刚烷酰氯也都可耐受. 通过一系列实验包括自由基捕获实验, Stern Volmer猝灭实验, 开关实验和18O标记实验, 提出了涉及铜配合物的光激发双自由基偶合反应机制. 光激发CuI-BINAP复合物A产生激发的*[CuI- BINAP]复合物B, 其与芳酰氯单电子转移形成铜复合物C, 并释放芳酰基自由基. 同时, 光激发硝基芳烃形成一个三重态双自由基中间体D, 酰基自由基可捕获该双自由基物种, 产生短寿命的自由基中间体E, 分解后释放出亚硝基苯F和羧酸氧自由基. 亚硝基苯是一种有效的自由基捕获剂, 可与第二个酰基自由基反应, 形成长寿命自由基G, G继续与另一酰基自由基重组产生双酰化中间体H, 其通过1,3-酰氧迁移并互变异构以提供所需的产物.
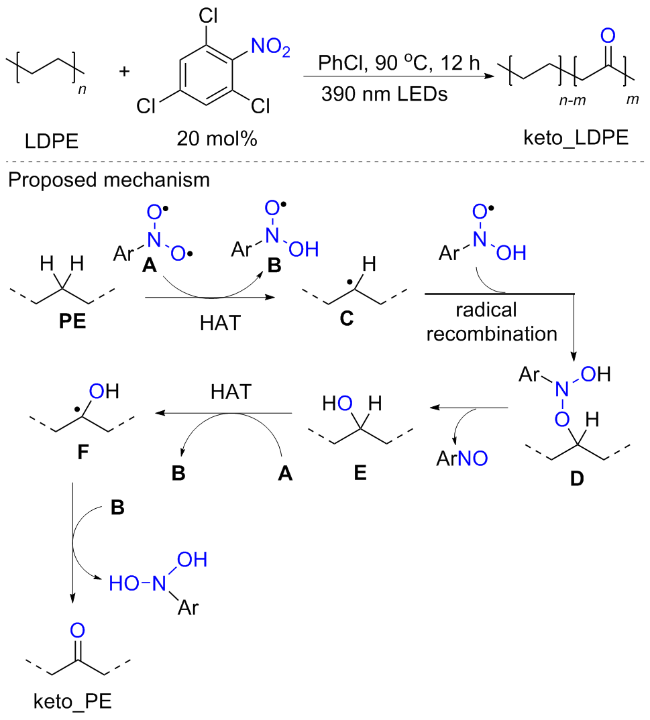
聚烯烃是应用最广泛的塑料, 由于其多功能性、耐用性和成本效益, 广泛应用于汽车零部件、纺织品和医疗设备等领域, 但是其抗降解性也加剧了环境保护的压力, 如何实现降解高分子的合成一直备受关注. 而含内酮的聚乙烯材料在保留其原有特性外, 还可增强附着力及亲水性, 这为后续降解提供了可能. 最近, Miyake 等[80]实现了一种光诱导硝基化合物作为氧源实现聚合物的羰基化修饰新方法(Scheme 39). 作者以低密度聚乙烯(LDPE)为底物, 发现2,4,6-三氯硝基苯的催化效果最好, 新戊醇和氯苯作为共溶剂对于产率的提升也较为重要. 基于相关实验研究, 作者提出反应机制可能为: 在光照下硝基芳烃形成三重态双自由基态, 该三重双自由基中间体与PE的C(sp3)—H键发生HAT反应产生烷基自由基中间体C和氧中心的二羟基苯胺自由基B; 随后, B和C的自由基重组导致中间体D; 接着D的裂解导致氧原子转移, 生成羟基中间体E和亚硝基苯副产物. 随后进一步经过HAT以产生α-羟基自由基中间体F; 最后F形成相应的酮产物和二羟胺副产物.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 总结
综上所述, 由于含氮化合物在医药、农业、化工和材料等领域有着广泛的应用, 以硝基芳烃作为潜在的氨源实现C—N键的偶联是目前的研究热点之一. 近些年, 通过自由基途径介导的硝基芳烃活化研究已取得重大进展. 相较于传统离子途径, 基于硝基自由基的偶联方案极大地丰富有机合成方法论, 特别是硝基的氧转移反应, 这些方法论为药物化学和药物的发展做出了巨大的贡献. 根据合成产物的类型不同, 大致分为自由基介导的烷胺或芳胺的合成、酰胺及其衍生物的合成、杂环的合成, 以及氧转移构建C—OH或C=O化合物. 详细讨论了反应底物的相容性、机理、应用、发展前景以及该领域的局限性. 其实除了硝基芳烃的硝基官能团活化, 近年来基于自由基介导的硝基烷烃活化[81]、自由基介导的硝基芳烃对位C(sp2)—H活化[82]、自由基介导的硝基芳烃C—NO2活化[83]也得到初步发展, 这些方法进一步拓展了硝基化合物在自由基偶合方面的应用.
尽管如此, 硝基自由基在交叉偶联中的应用仍然不够成熟, 特别是当它被用作SET或HAT激活其他惰性偶联伙伴的中间体时, 为未来的突破留下了巨大的潜力. (1)目前已实现的偶联大多限于烯烃类化合物. (2)对于卤素需要活化更强的C—Cl键或C—Br键(而不是C—I键, C—I键其实是更容易均裂产生自由基的). 我们期望更多类型的底物能被用来与硝基偶合, 特别是那些容易作为自由基受体或供体的化合物. (3)上述反应大多局限于光催化或过渡金属协同光催化实现硝基自由基的产生, 使得亚硝基芳烃几乎是所有反应的关键中间体, 尝试更多的电化学或者与有机催化剂配合使用的无金属方法, 这些战略可能扩展更复杂的反应系统并涉及改变硝基自由基过程的反应性和选择性. 这些用于生成硝基自由基中间体的新方案也将导致对难以捉摸的物种进行更严格的研究和更深层的理解和量化, 并为设计利用硝基自由基独特合成潜力的创新转化铺平道路.
(Lu, Y.)