


1 结果与讨论
1.1 单、双室乙醇胺电氧化反应的研究
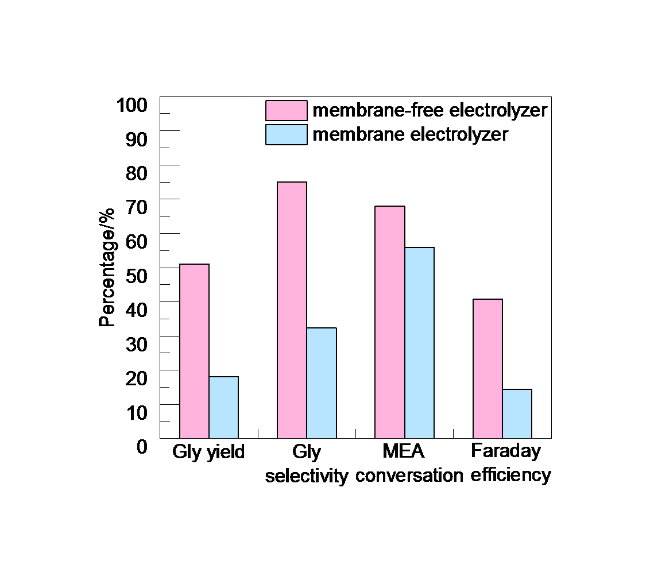
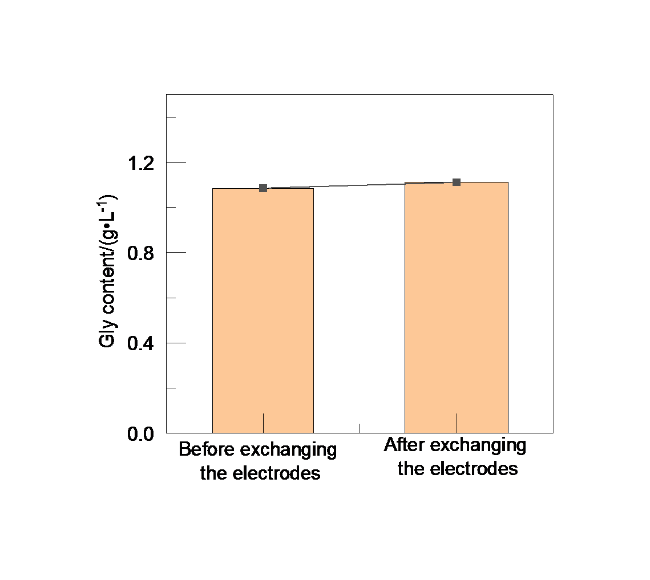
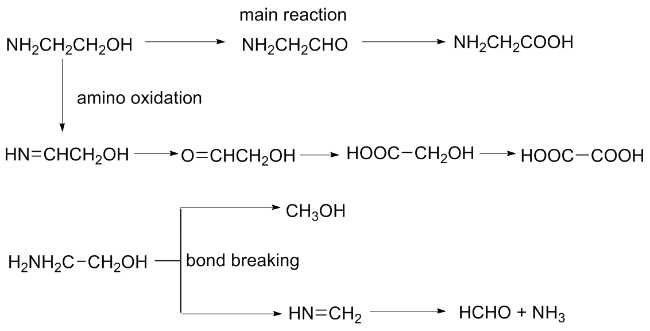
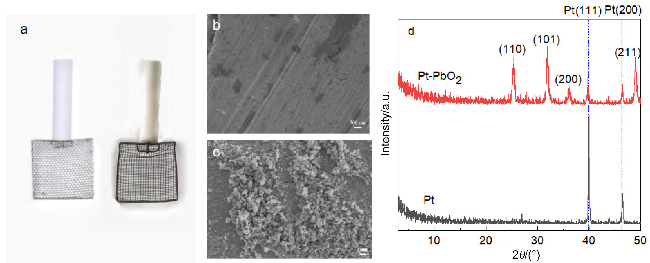
图3 (a)铅作为阴极时无隔膜电解前后铂阳极的对比图; (b)无隔膜电解前的Pt电极的SEM图和(c)无隔膜电解后的Pt电极的SEM图; (d)无隔膜电解前后的Pt电极的XRD分析图Figure 3 (a) Comparison diagram of the Pt anode before and after undivided electrolysis with lead as the cathode; (b) SEM image of the Pt electrode before undivided electrolysis and (c) SEM image of the Pt electrode after undivided electrolysis; (d) XRD patterns of the Pt electrode before and after undivided electrolysis |
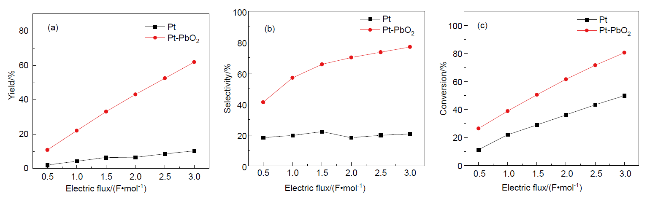
表1 Pt、PbO2和Pt-PbO2对Gly收率、Gly选择性、MEA转化率和法拉第效率的影响Table 1 Influence of Pt, PbO2 and Pt-PbO2 on the yield of Gly, selectivity of Gly, MEA conversion, and Faraday efficiency |
| Anode | Gly yield/% | Gly selectivity/% | MEA conversion/% | Faraday efficiency/% |
|---|---|---|---|---|
| Pt | 27.02 | 45.40 | 59.52 | 21.62 |
| PbO2 | 70.28 | 78.50 | 89.54 | 56.25 |
| Pt-PbO2 | 80.99 | 81.45 | 99.43 | 64.81 |
1.2 Pt与Pt-PbO2电氧化乙醇胺机理研究
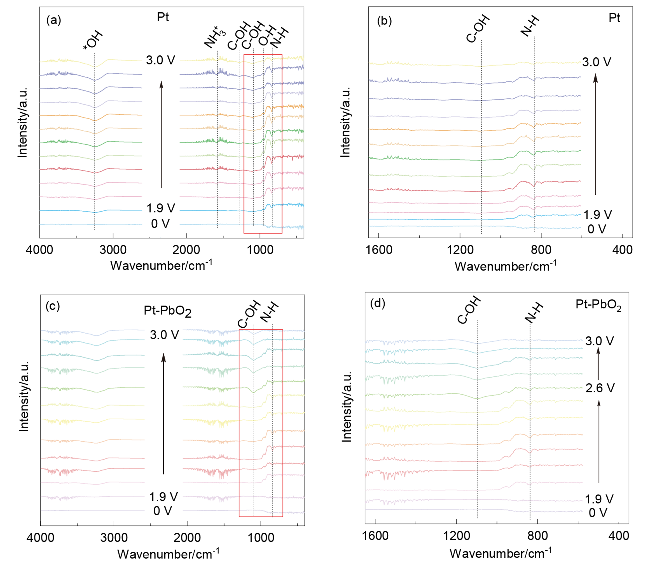
图5 电位为1.9~3.0 V时Pt电极(a, b)和Pt-PbO2电极(c, d)在0.3 mol/L MEA+2.0 mol/L H2SO4溶液中的原位多步阶跃红外光谱图(vs. Ag/AgCl) (图b、d分别为图a、c中对应红框中的放大图)Figure 5 In-situ multi-step staircase infrared spectroscopy of Pt electrode (a, b) and Pt-PbO2 electrode (c, d) in 0.3 mol/L MEA+2.0 mol/L H2SO4 solution at the research potential of 1.9~3.0 V (vs. Ag/AgCl) (b and d are the enlarged view of Figures a and c) |
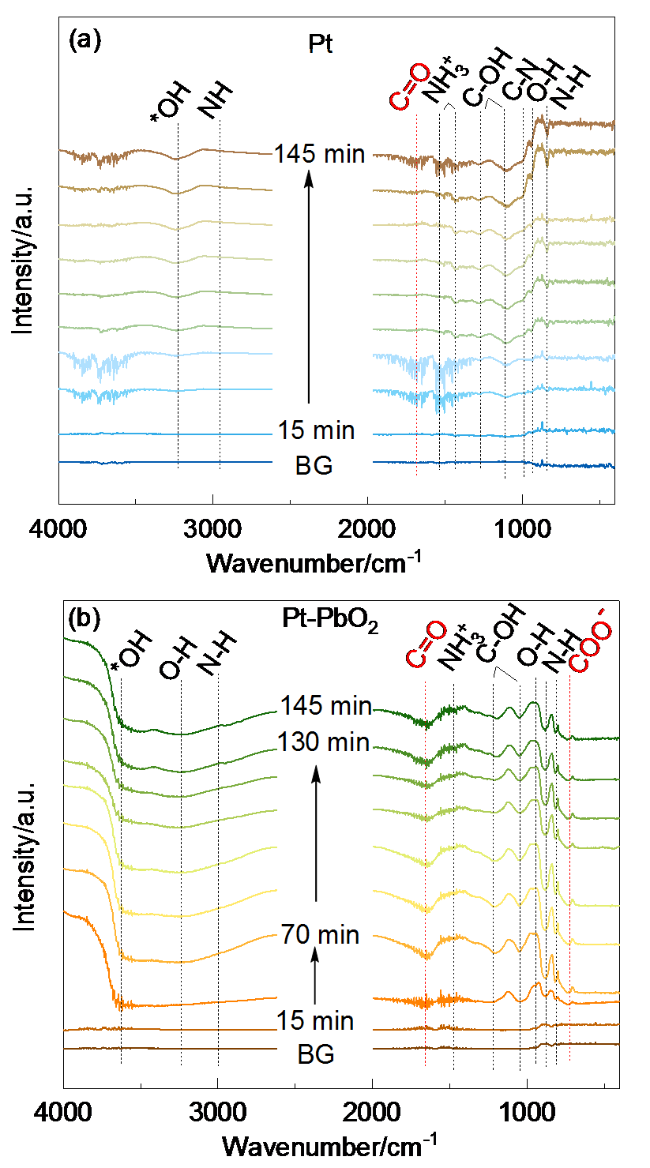
1.3 电解条件的优化
1.3.1 酸碱环境的影响
表2 酸、碱环境对MEA电合成Gly的影响Table 2 Effect of acid-base environment on MEA electrosynthesis of Gly |
| Acid-base environment | Gly yield/% | Gly selecti- vity/% | MEA con- version/% |
|---|---|---|---|
| Acidic media | 79.22 | 88.61 | 89.40 |
| Alkaline media | 3.09 | 5.25 | 58.99 |
1.3.2 酸种类的优化
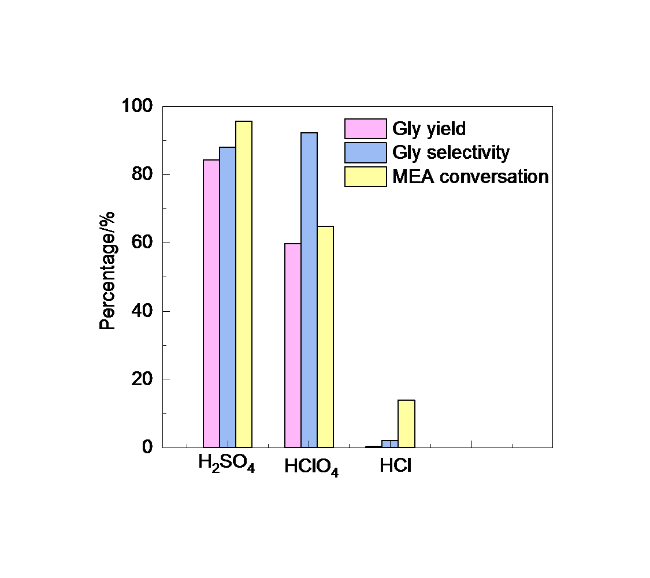
1.3.3 酸浓度的优化
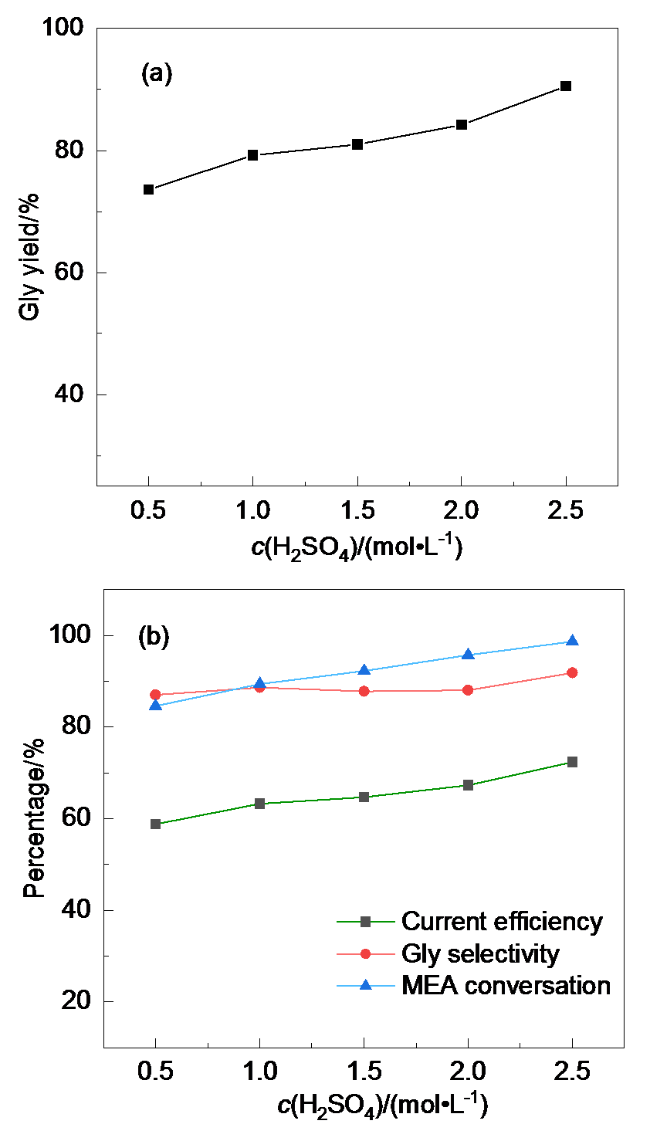
1.3.4 乙醇胺浓度的优化
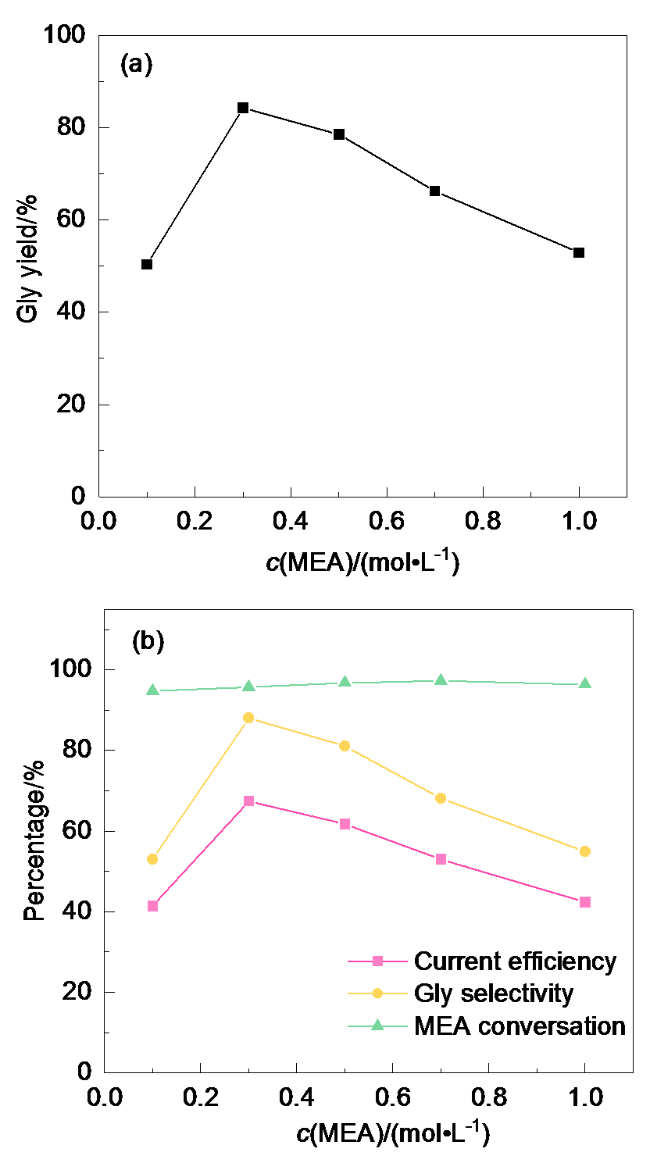
1.3.5 电流密度的调控
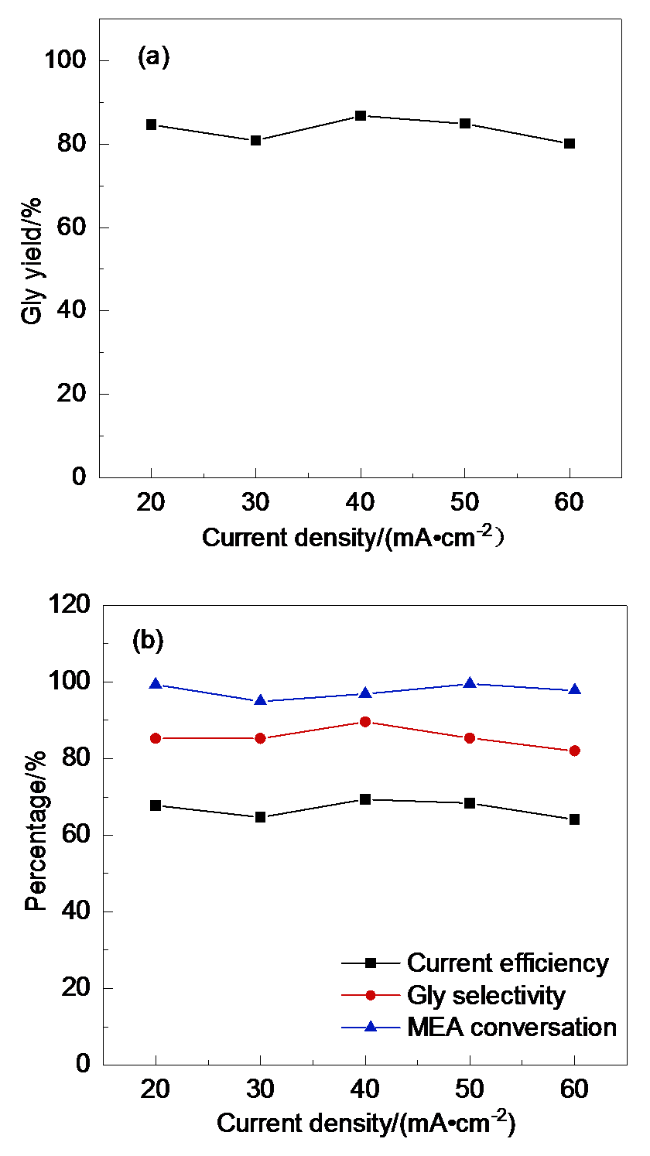
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1.3.6 反应的重复性
表3 电解反应重复性测试结果Table 3 Electrolysis reaction repeatability test results. |
| Serial number | Gly yield/% | Gly selecti- vity/% | MEA con- version/% | Faraday efficiency/% |
|---|---|---|---|---|
| 1 | 84.23 | 88.03 | 95.69 | 67.28 |
| 2 | 86.30 | 90.16 | 95.73 | 69.53 |
| 3 | 85.57 | 88.43 | 96.77 | 68.50 |