


季碳中心(包含全碳季碳中心和杂原子取代的季碳中心)作为一类重要的结构单元, 广泛存在于天然产物及功能分子的核心骨架中, 并且其对分子结构复杂程度的贡献也是显而易见的(图1). 然而, 由于季碳中心本身所具有的复杂的化学结构及显著的空间位阻效应, 使得这类结构单元的合成充满了巨大的挑战, 因此, 开发快速高效构筑季碳中心的合成方法学, 对于复杂天然产物及功能分子核心骨架的构筑及对应天然产物的全合成是至关重要的. 钯催化串联反应在复杂天然产物的合成中已经得到了非常广泛的应用[1], 其中, 通过该串联反应以一步转化实现含季碳中心多环体系的构建可以高效实现复杂分子中核心单元的快速合成, 进而缩短合成步骤, 提高合成效率. 而该串联反应的优势主要表现在以下三个方面: (1)反应通常由sp2杂化的烯基卤代物启动, 其相对较小的空间位阻有利于季碳中心的引入; (2)反应过程中所生成的C—Pd(II)键可以经过多次的连续迁移插入形成多根C—C键与C—X键, 从而实现复杂环系的构筑; (3)反应过程中官能团兼容性良好, 便于该转化在复杂体系中的应用.
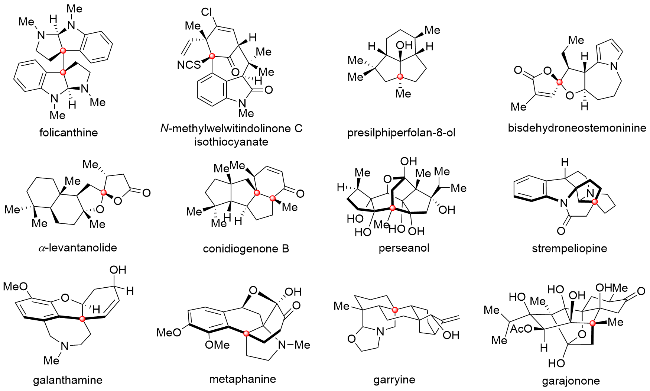
鉴于钯催化串联环化反应在天然产物全合成中的实用性, 为了更好地理解该串联反应的具体反应过程, 本文我们将结合近十年来所发展的有代表性的合成实例对钯催化串联反应在合成含季碳中心复杂天然产物中的应用进行简要阐述.
1 六氢吡咯吲哚生物碱的全合成
1.1 六氢吡咯吲哚生物碱(+)-esermethole (6a)和(+)-physostigmine(6b)的全合成
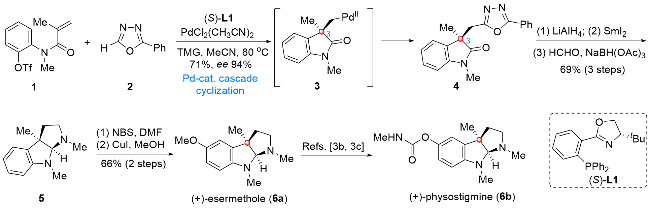
六氢吡咯吲哚生物碱是吲哚生物碱家族中一类重要的组成单元, 其表现出丰富的分子结构多样性和良好的生物活性[2]. 作为该家族生物碱中结构最为简单的分子之一, (+)-esermethole (6a)具有顺式稠合的苯并氮杂5/5环系, 有一个全碳季碳中心, 其合成难点主要集中于上述结构单元的构建. 2015年, 祝介平课题组[3a]发展了一种以N-芳基丙烯酰胺作为底物的不对称钯催化串联环化反应, 通过该转化一步实现C(3)位季碳取代氧化吲哚结构的构建(Scheme 1). 化合物1与2在Pd(CH3- CN)2Cl2作为钯源、(S)-L1作为手性配体的条件下可以经中间体3顺利发生不对称串联环化反应, 得到化合物4, 后续经过氧化态及取代基的调整, 经中间体5最终实现(+)-esermethole (6a)的全合成及(+)-physostigmine (6b)的形式全合成.
1.2 六氢吡咯吲哚生物碱(+)-physovenine (11), (+)-physostigmine (12), (+)-phenserine (13)及二聚体(+)-folicanthine (15)的合成
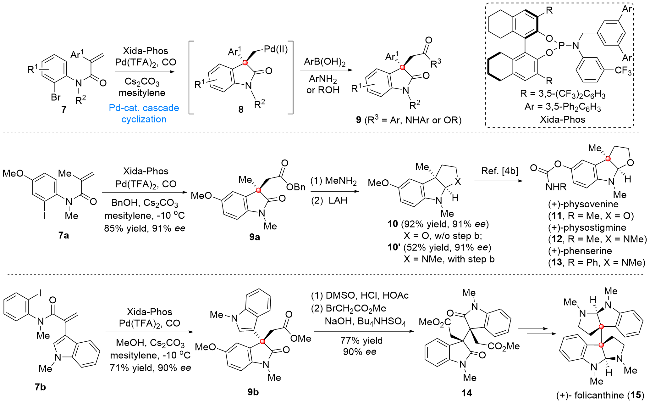
2020年, 关正辉课题组[4a]发展了基于N-芳基丙烯酰胺底物的不对称钯催化插羰串联环化方法学, 后期通过该串联反应为关键步骤, 成功地实现了六氢吡咯吲哚生物碱(+)-physovenine (11), (+)-physostigmine (12), (+)-phenserine (13)及二聚体(+)-folicanthine (15)的合成(Scheme 2). 作者在其所发展的Pd(TFA)2和Xida-Phos分别作为钯源和手性配体的条件下, 成功实现了酰胺7参与的不对称钯催化串联环化过程, 所生成的中间体8经羰基插入及后续不同类型试剂参与的还原消除过程, 得到一系列C(3)位不同取代的手性氧化吲哚类化合物9. 在此方法学的基础上, 作者以7a作为环化前体, 通过上述串联过程, 以85%收率及91% ee值成功实现了化合物9a的合成. 随后, 化合物9a分别经简单的还原环化及MeNH2胺解/还原环化反应, 分别得到高级中间体10和10', 后期通过文献报道的转化过程[4b], 实现了六氢吡咯吲哚生物碱(+)-physovenine (11), (+)-physo- stigmine (12)和(+)-phenserine (13)的形式合成. 接下来, 作者尝试了化合物7b参与的串联环化过程, 并以71%收率及90% ee值得到化合物9b, 后期经氧化和烷基化过程, 得到高级中间体14, 化合物14经过MeNH2胺解及后续还原环化过程, 最终实现二聚体(+)-folicanthine (15)的不对称全合成.
2 氧化吲哚生物碱(-)‑N‑methylwelwitindo- linone C isothiocyanate (24)的全合成
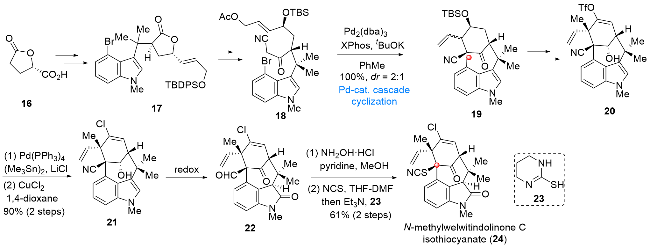
氧化吲哚生物碱(-)‑N‑methylwelwitindolinone C isothiocyanate (24)是由Moore课题组[5]于1994年从软管藻属蓝藻中分离得到的氧化吲哚类天然产物, 其生物活性主要表现在对人类癌细胞具有显著的抑制作用. 从结构上分析该分子具有含氧化吲哚结构的多取代双环[4,3,1]癸烷骨架, 其中包含一个桥头季碳中心, 合成上存在极大的挑战性, 因此, 实现含桥头季碳中心的多取代双环[4,3,1]癸烷骨架的构建就成为能否实现该分子全合成的关键. 2015年, Hatakeyama课题组[6]发展了以溴代吲哚18作为底物的钯催化串联环化反应, 通过该串联反应一步实现上述含桥头季碳中心的双环[4,3,1]癸烷骨架的构建, 在此基础上, 最终完成氧化吲哚生物碱(-)‑N‑methylwelwitindolinone C isothiocyanate (24)的全合成(Scheme 3). 从手性化合物16出发, 经碳链延长和吲哚片段的引入得到化合物17, 后经内酯基团的转化, 所得化合物18在Pd2(dba)3/XPhos/tBuOK的作用下经烯醇负离子中间体顺利发生烯丙基化/烷基化串联反应, 以定量的收率得到具有双环[4,3,1]癸烷骨架的化合物19, 一步实现了分子骨架的构建. 在此基础上, 化合物19经烯醇三氟甲磺酸酯基团和甲基的引入, 转化为化合物20. 接下来, 往化合物20中引入氯原子, 所得中间体21经过不同位点氧化态的调整, 得到化合物22. 向化合物22引入硫氰基基团, 最终实现氧化吲哚生物碱(-)‑N‑methylwelwitindolinone C isothiocyanate (24)的全合成.
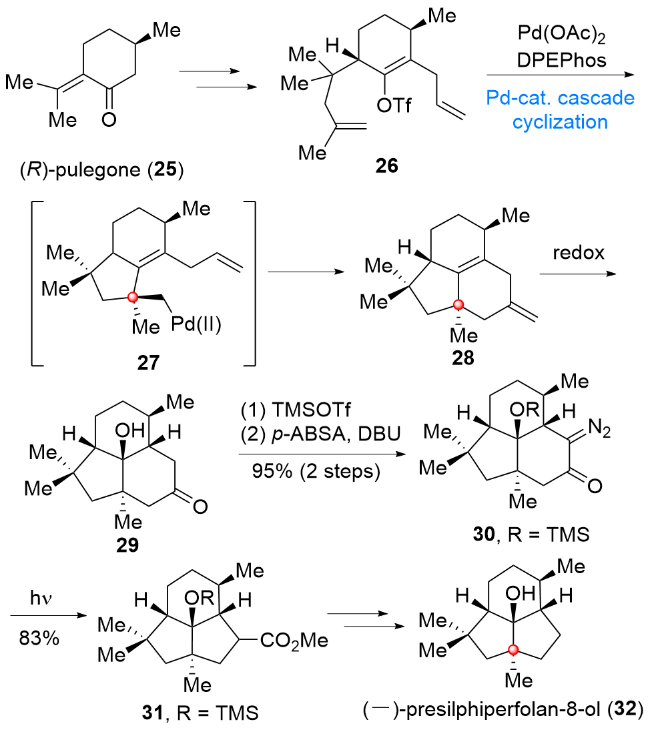
3 倍半萜类天然产物(-)-presilphiperfolan- 8-ol (32)的全合成
天然产物(-)-presilphiperfolan-8-ol (32)是由Bohl- mann课题组[7]于1981年从绵叶菊属植物中分离得到的倍半萜类化合物. 从结构上看, 该分子具有[6,5,5]三环体系, 其中包含一个具有高张力的反式稠合[3,3,0]辛烷结构及两个连续的季碳中心, 这些结构特点使得该分子表现出极大的合成难度. 2017年, Snyder课题组[8]针对该分子的结构特点, 设计了一条巧妙的合成路线, 其核心合成策略体现在, 首先通过分子内钯催化串联环化反应及后续环氧化反应完成含双季碳中心的反式[4,3,0]壬烷骨架的构建, 后期利用Wolff重排反应实现由张力较小的反式[4,3,0]壬烷骨架向张力较大的反式3,3,0]辛烷骨架的转化, 在此基础上, 最终实现该天然产物的全合成(Scheme 4). 从商业获得的化合物(R)-pulegone (25)出发, 经双烯丙基侧链及三氟甲磺酸酯的引入, 得到环化前体26. 在Pd(OAc)2/DPEPhos作用下, 化合物26可以经中间体27顺利发生分子内串联环化反应, 得到三环化合物28, 然后经氧化态的调整, 即可得到具有反式[4,3,0]壬烷骨架的化合物29. 化合物29经重氮基团的引入, 得到重氮化合物30, 30在光照条件下顺利经过Wolff重排发生缩环反应, 得到具有高张力反式[3,3,0]辛烷骨架的化合物31. 最后, 化合物31经甲酯基团和相应保护基的脱除, 实现(-)-presilphiperfolan-8-ol (32)的首次不对称全合成.
4 氧杂螺内酯类天然产物的全合成
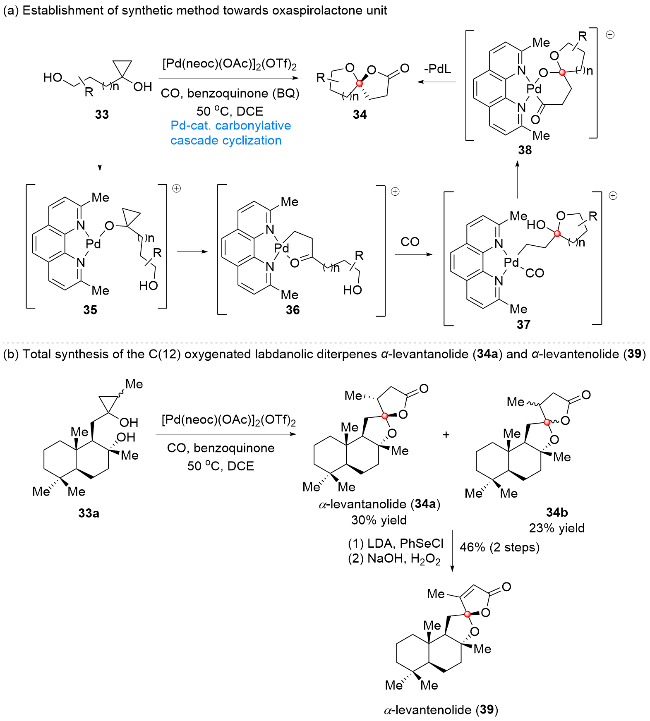
4.1 氧杂螺内酯结构单元合成方法学的建立及其在C(12)位氧杂半日花烷型二萜合成中的应用
2016年, 代明骥课题组[9]发展了以γ/δ-羟基环丙醇作为底物合成含氧杂螺内酯单元化合物的钯催化插羰串联环化反应的方法学, 并以此串联反应为关键步骤, 实现了氧杂半日花烷型二萜α-levantanolide (34a)和α- levantenolide (39)的合成(Scheme 5). 以γ/δ-羟基环丙醇33作为底物, 经过系统的条件筛选, 作者发现该串联过程在一氧化碳气氛下以[Pd(neoc)(OAc)]2(OTf)2作为钯源、苯醌作为氧化剂、二氯乙烷(DCE)作为溶剂时, 可以顺利发生, 以中等到优秀的产率得到一系列含不同环系及取代基的氧杂螺内酯化合物34. 同时, 利用高分辨率电喷雾电离质谱(ESI-MS)技术, 作者对该串联反应的机理进行了探究, 并在此基础上提出了一种可能的转化过程. 首先, 底物和钯催化剂作用形成中间体35, 后经β位C—C键断裂及后续环化过程生成中间体36, 在CO作用下, 中间体36发生羰基插入生成中间体37, 后经螺环缩酮的形成得到中间体38. 最后, 化合物38经还原消除得到氧杂螺内酯产物34, 该过程中生成的Pd(0)中间体在苯醌作用下可氧化为Pd(II)中间体, 并重新参与到上述催化过程中. 在完成上述方法学的建立后, 作者将该方法学应用于C(12)位氧杂半日花烷型二萜的合成. 如Scheme 5所示, 以化合物33a为底物, 通过上述串联环化过程可以直接实现天然产物α-levantanolide (34a)的合成, 同时伴随有其差向异构体34b的生成. 接下来, 经酯羰基α位苯硒基的引入及后续消除反应, 化合物34a与34b均可转化为天然产物α-levantenolide (39), 从而实现了该家族天然产物的简洁高效全合成, 同时也证明了该方法学的实用性和高效性.
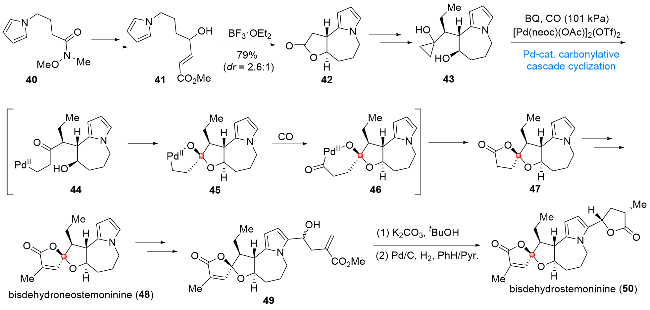
4.2 百部生物碱bisdehydroneostemoninine (48)和bisdehydrostemoninine (50)的全合成
百部生物碱bisdehydroneostemoninine (48)和bisde- hydrostemoninine (50)是由叶阳课题组[10]于2006年从百部科植物中分离得到的两个天然产物, 从结构上分析, 该类分子具有[5,5,7,5]四环基本骨架, 其中包含一个芳香性的吡咯环片段、一个敏感的氧杂螺环内酯单元以及四个连续的立体中心. 上述结构特点严重地制约了这类分子在合成方面的研究进展. 其中, 不稳定氧杂螺环内酯单元的高效构建是这类分子在合成方面取得突破的关键所在. 2016年, 代明骥课题组[11]发展了一种通过钯催化插羰串联环化反应高效构建氧杂螺环内酯单元的合成方法学, 并成功将其应用于上述两个百部生物碱的全合成中(Scheme 6). 由化合物40出发, 经碳链延长、分子内Friedel-Craft反应和乙基及环丙醇片段的引入, 即可得到串联环化前体43. 在Pd(II)作用下, 环丙醇43首先发生C—C键的断裂得到中间体44. 接下来, 该中间体转化为缩酮中间体45后发生CO的插入, 所形成的酰基钯中间体46经还原消除得到螺环缩酮产物47. 化合物47经甲基基团的引入及氧化态的调整, 实现了天然产物bisdehydroneostemoninine (48)的全合成, 后期引入γ-丁内酯片段后, 最终完成天然产物bisdehydroste- moninine (50)的全合成.
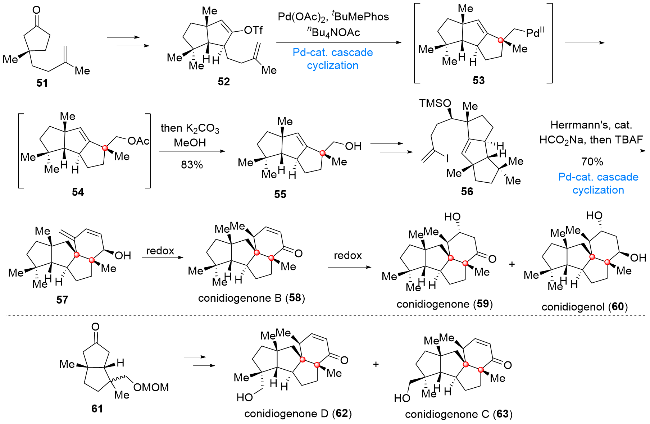
5 Cyclopianes二萜的全合成
Cyclopianes二萜是从青霉菌的发酵液中分离得到的一类次生代谢产物[12], 从结构上看, 这类天然产物具有[5,5,6,5]四环基本骨架, 包含多个连续的手性中心以及全碳季碳中心, 尤为特别的是, 该家族分子中所含官能团及杂原子的种类和数目都是非常有限的, 上述结构特点使得这类二萜的合成充满了巨大的挑战性. 2019年, Snyder课题组[13]提出了一种基于季碳中心导向的合成策略. 在该合成策略的指导下, 作者采用钯催化环化反应作为关键步骤, 成功实现了一系列cyclopianes二萜的全合成(Scheme 7). 从化合物51出发, 经若干官能团转化得到化合物52, 在Pd(OAc)2/tBuMePhos/ nBu4NOAc的作用下, 化合物52发生分子内Heck环化反应, 依次经中间体53和54得到三环产物55, 接下来向化合物55中引入高烯丙基侧链, 所得化合物56在Herrmann催化剂/HCO2Na的反应体系中, 通过还原Heck反应再次发生环化, 得到四环产物57. 在该化合物的基础上, 仅需通过简单的氧化态调整, 即可最终实现conidiogenone B (58)、conidiogenone (59)和conidiogenol (60)的全合成. 而由底物61出发, 经过与之前相同的合成策略, 同样可以完成天然产物conidiogenones C (62)和D (63)的全合成.
6 二萜类天然产物(+)-perseanol (72)的全合成
天然产物(+)-perseanol (72)是由Fraga课题组[14]于1996年从加那利群岛的灌木Persea indica中分离得到的一种二萜类化合物, 生物活性研究表明该分子对鳞翅目害虫表现出很高的拒食活性及对哺乳动物更小的毒性作用. 从结构上分析, (+)-perseanol (72)具有高度稠和的环系骨架, 其中包含一个桥连的七元环片段和敏感的半缩酮结构、两组顺式稠和的邻二羟基单元以及多个氧杂及全碳季碳中心. 上述结构特点使得该分子的合成不仅在合成策略的设计上充满了前所未有的挑战性, 而且在合成效率的提升上表现出极大的难度. 2019年, Reisman课题组[15]发展了通过钯催化插羰串联环化反应一步实现(+)-perseanol (72)中内酯桥环单元构建的合成策略, 在此基础上, 仅通过16步转化实现该分子的首次全合成(Scheme 8). 从醛64和碘代物65出发, 在nBuLi作用下经1,2-加成反应使两片段连接, 得到环化前体66. 化合物66通过分子内Heck型环化(中间体67)、CO迁移插入(中间体68)和酰基钯中间体的还原消除等转化过程, 得到五环产物69, 一步实现内酯桥环单元及全碳季碳中心的构筑. 接下来, 通过后期官能团转化得到环氧化合物70, 在LiPhNap作用下, 化合物70经酯羰基启动的分子内还原环化反应, 顺利实现了四氢呋喃(THF)环系的构建, 得到环化产物71. 最后, 经过简单的保护基脱除, 实现(+)-perseanol (72)的首次全合成.
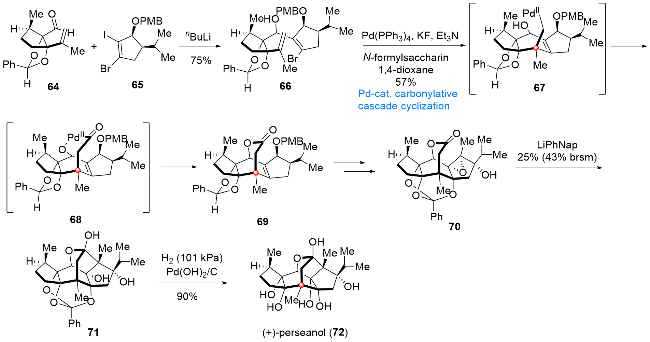
7 单萜吲哚类生物碱(+)-strempeliopine的全合成
单萜吲哚类生物碱schizozygane主要是从非洲Schizozygia caffaeoides属植物中分离得到的一类天然产物, 生物学研究表明该类天然产物具有抗真菌和抗疟原虫等生物活性[16]. 从结构上看, 该家族分子具有复杂的[6,5,6,6,5,6]六环基本骨架, 包含4~6个连续的手性中心, 其中两个分别为全碳季碳中心和氮杂季碳中心, 上述结构特点使得针对这类分子的合成面临着巨大的挑战, 也为其集群式合成提供了良好的机遇. 2021年, 张敏课题组[17]发展了一种以吲哚去芳化环化和钯催化分子内Heck型插羰串联环化反应为关键步骤的合成策略, 在该合成策略的指导下, 成功实现了schizozygane生物碱的集群式合成(Scheme 9). 从色胺73出发, 经环丙醇片段的引入、Bischler-Napieralski环化反应及后续保护基和氧化态的调整, 可以得到环化前体74, 在Fe(NO3)3作用下, 化合物74经氧化去芳化/分子内环化过程, 可以顺利转化为四环产物75. 接下来, 经过氧化态的调整和碳链的延长, 得到烯基碘代物76, 76在Pd(OAc)2/CO作用下可以顺利经中间体77和78发生分子内Heck型插羰串联环化反应, 得到六环化合物79, 一步完成天然产物骨架的构建, 后经双键的还原, 即可实现(+)- strempeliopine (80)的全合成. 而由底物81出发, 经过与前述相同的转化过程, 最终可以实现该家族天然产物的集群式合成.
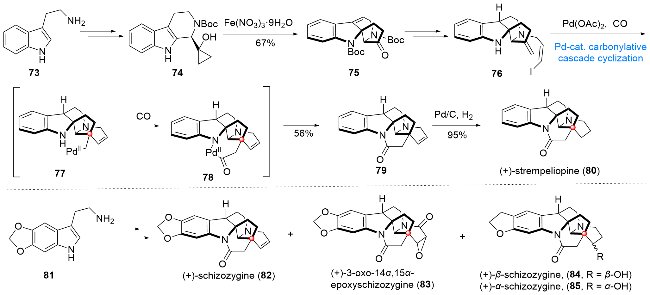
8 石蒜科生物碱galanthamine (94)和lyco- ramine (95)的全合成
石蒜科生物碱galanthamine (94)和lycoramine (95)是从石蒜科植物雪钟花中分离得到的一类具有相同环系组成的天然产物[18], 其生物活性主要表现在具有很强的抗乙酰胆碱酯酶(AChE)作用, 该天然产物的氢溴酸盐已被作为商业化的药品(商品名: Radazyne)用于阿尔茨海默症的治疗. 从结构上看, 该类分子具有复杂的四环骨架, 包括苯并呋喃环片、顺式稠合的5/6并环体系以及一个氮杂七元环单元, 同时包含一个全碳季碳中心, 上述结构特征导致该分子在合成上具有一定的挑战性. 2021年, 赵玉明课题组[19]发展了一种以钯催化插羰串联环化反应和分子骨架重整为关键步骤的合成策略, 在该合成策略的指导下, 成功实现了上述两个生物碱的发散式合成(Scheme 10). 从商业可得的化合物86出发, 经简单的偶联及官能团调整, 即可得到环化前体88. 在Pd(OAc)2/AgOTf/2,6-二叔丁基吡啶(DTBP)/CO作用下, 化合物88可以顺利发生作者所设计的串联环化反应, 依次经过中间体89和90得到四环产物91, 然后经氨解及分子内氧化环化反应, 得到氮杂半缩醛92. 在BF3•Et2O作用下, 92经骨架重整顺利转化为93, 实现目标天然产物中分子骨架的构建, 经过氧化态的调整, 最终实现galanthamine (94)和lycoramine (95)的全合成.
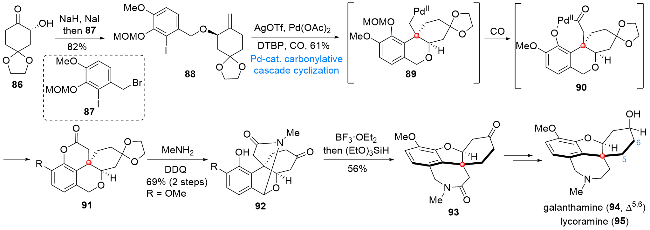
9 莲花烷生物碱metaphanine (104)和oxo- epistephamiersine (105)的全合成
莲花烷生物碱是从Stephania属植物中分离得到的一大类次生代谢产物, 这类天然产物在抗病毒及抗菌等方面表现出良好的生物活性[20]. 作为其中的代表性分子, metaphanine (104)和oxoepistephamiersine (105)在结构上具有与众不同的氮杂[4,4,3]螺桨烷骨架以及包含敏感半缩酮基团的四氢呋喃环单元, 上述结构特点在展现出该类分子较大合成难度的同时, 也成为吸引有机合成化学家的主要关注点之一. 2023年, 赵玉明课题组[21]设计了以钯催化插羰串联环化反应和氧化环化反应为关键步骤的合成路线, 利用该合成路线实现了上述莲花烷生物碱的发散式合成(Scheme 11). 由商业可得的原料96出发, 经两片段偶联及简单官能团化, 得到环化前体97. 在Pd(OAc)2/PPh3/AgOTf/DTBP/CO作用下, 97可以顺利发生串联环化反应, 依次经过中间体98和99得到四环产物100, 然后经氧化态的调整得到烯酮化合物101, 101经氮原子的引入得到酰胺102, 102在二甲基过氧化酮(DMDO)和硝酸铈铵(CAN)作为氧化剂的条件下发生连续氧化反应, 顺利构筑四氢呋喃环并引入半缩酮单元得到五环产物103, 经过氧化态的调整, 最终可以实现metaphanine (104)和oxoepistephamiersine (105)的发散式全合成.
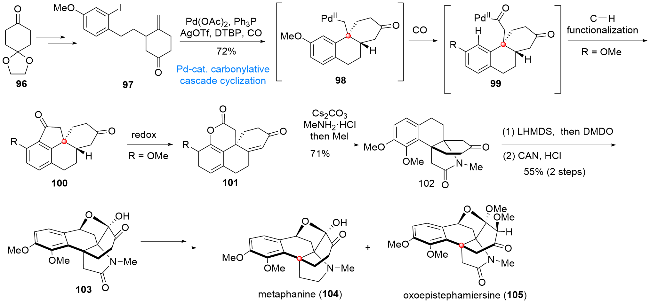
10 二萜类生物碱(-)-garryine (111)的全合成
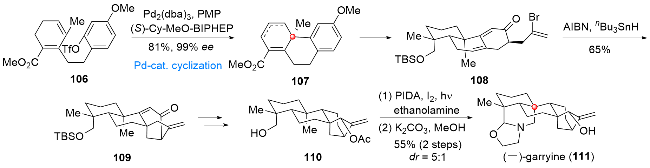
Veatchine型C20二萜类生物碱主要是从garrya属植物中分离得到一类结构复杂且生物活性丰富的天然产物[22], 作为其中的代表性分子, (-)-garryine (111)在结构上具有复杂的[6,6,6,6,5,5]六环环系骨架, 其中包含稠合的哌啶-噁唑烷酮结构、[3,2,1]桥环单元以及三个全碳季碳中心, 如何高效且简洁地完成该分子的合成, 成为有机合成领域亟待解决的一个难题. 2023年, 秦勇课题组[23]发展了一种以钯催化环化反应和分子内自由基环化反应为关键步骤的合成策略, 利用该合成策略高效地完成了(-)-garryine (111)的全合成(Scheme 12). 化合物106在钯催化条件下经分子内Heck型环化反应得到三环产物107, 后经氧化态的调整及烯丙基侧链的引入得到烯基溴108. 在偶氮二异丁腈(AIBN)/nBu3SnH作用下, 108可以顺利通过分子内自由基环化反应构筑[3,2,1]桥环单元, 得到四环产物109. 接下来, 经过官能团转化, 所得伯醇110在光照条件下经碘苯二乙酸(PIDA)氧化/乙醇胺缩合及环化过程, 最终完成(-)-garryine (111)的全合成.
11 天然产物euphorbialoid A (119)的全合成
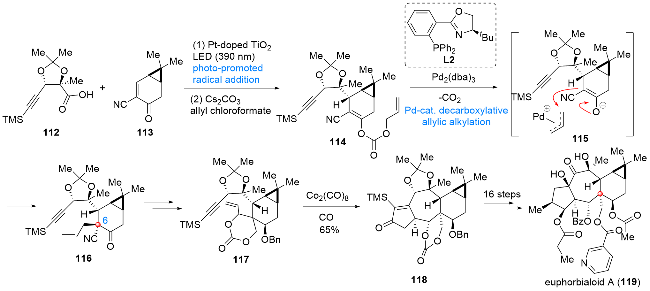
Euphorbialoid类化合物是从大戟科植物土瓜狼毒中分离得到的一类具有反式稠和[5,7,6]三环骨架的二萜类天然产物. 作为该家族天然产物的代表性分子, euphorbialoid A (119)在上述三环基本骨架的基础上不仅增添了环丙烷单元, 而且在(C3)、C(5)、C(7)及C(17)位分别引入了不同结构的酯基官能团, 从而增加了该分子结构上的复杂性以及合成上的挑战性[24]. 2024年, Inoue课题组[25]发展了一条以钯催化脱羧烯丙基化及Pauson- Khand反应作为核心步骤的合成路线, 在此基础上实现了euphorbialoid A (119)的首次全合成(Scheme 13). 作者以底物112和113作为起始原料, 经光促进的自由基加成反应实现两片段连接, 后续经烯丙基碳酸酯化得到114. 在Pd2(dba)3作用下, 底物114经中间体115发生脱羧烯丙基化反应, 构筑C(6)位季碳中心, 得到化合物116. 接下来, 经过官能团及保护基的调整, 所得烯炔化合物117在Co2(CO)8作用下发生Pauson-Khand反应, 实现了关键的[7,5]并环结构单元的构建, 得到化合物118. 后续经过不同位点氧化态的调整及多种类型侧链酯基基团的引入, 作者成功实现了euphorbialoid A (119)的首次全合成.
12 尼亚那属二萜garajonone (129)的全合成
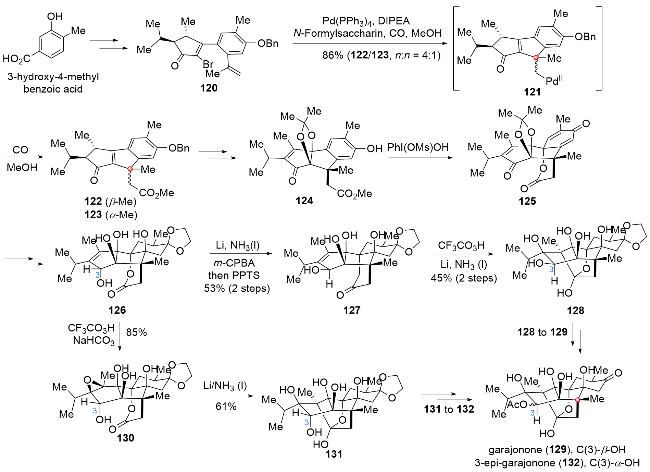
尼亚那属二萜是从中美洲和南美洲大枫子科灌木中分离得到的一类具有复杂结构和显著生物活性的天然产物. 从结构上看, 该类分子具有[6,6,5,5,5]五环基本骨架和8个连续的含氧原子立体中心, 使得该家族分子成为目前文献报道的氧化态最高的一类复杂天然产物. 生物活性研究表明, 该类天然产物对兰尼碱受体(PyRs)表现出良好且专一的调节作用, 在治疗中枢神经系统疾病、心血管疾病以及免疫抑制活性等方面表现出潜在的应用价值[26]. 2024年, 赵玉明课题组[27]发展了包含钯催化串联环化/插羰酯化、氧化去芳构化/内酯化及还原环化等关键步骤的两阶段法合成策略, 在该合成策略的指导下成功实现了garajonone (129)和3-epi-garajonone (132)的首次全合成(Scheme 14). 作者从起始原料3-羟基-4-甲基苯甲酸出发, 经简单的转化过程得到溴代物120. 在Pd(PPh3)4、N,N-二异丙基乙胺(DIPEA)和N-For- mylsaccharin体系及一氧化碳气氛下, 底物120可以顺利地发生钯催化串联环化/插羰酯化过程, 经中间体121得到三环化合物122及其差向异构体123. 接下来, 经过氧化态和保护基的调整得到化合物124, 124在PhI(OMs)OH作用下发生氧化去芳构化/内酯化串联过程, 得到双烯酮产物125. 随后, 经过保护基的调整及对应位点氧化态的改变, 得到共同中间体126. 从中间体126出发, 一方面经Li/NH3(l)促进的还原环化、环氧化及酸性条件促进的碎裂化过程, 得到C(3)位构型正确的化合物127, 该化合物再经过环氧化及后续还原环化过程, 得到五环化合物128, 最后经过保护基团的调整, 即可实现garajonone (129)的首次全合成; 另一方面, 共同中间体126直接经环氧化及后续还原环化过程, 得到五环化合物131, 经过三步转化对保护基进行调整, 最终可以实现3-epi-garajonone (132)的首次全合成.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
13 结论
钯催化串联环化反应在构筑含季碳中心的复杂环系结构方面表现出良好的底物普适性以及反应条件的简洁性, 上述总结不仅让我们对该串联反应的底物与产物的结构特点、反应条件以及反应机理有了更深入的学习与理解, 而且也为其在更多天然产物及药物分子合成中的应用提供了一些参考与指导.
(Zhao, C.)