将喜树碱(30 mg, 0.086 mmol)分散于3 mL水中, 在冰浴下滴加0.76 mL浓硫酸, 使喜树碱完全溶解. 在室温下向体系中加入FeSO4•7H2O (42 mg, 0.15 mmol), 搅拌5 min后加入自由基源(1.29 mmol). 将体系移入冰浴, 滴加质量分数为30%的H2O2 (35 μL, 1.14 mmol), 过程中控制内温<20 ℃. 反应30 min后补加FeSO4•7H2O (42 mg, 0.15 mmol), 搅拌5 min后将体系移入冰浴, 滴加质量分数为30%的H2O2 (35 μL, 1.14 mmol), 过程中控制内温<20 ℃, 继续反应30 min. 反应结束后加入质量分数为30%的NaOH水溶液淬灭反应, 过滤析出的固体, 水相以乙酸乙酯(EA)萃取(5 mL×3), 浓缩, 除特别说明外, 所得粗产物均采用Flash柱纯化[二氯甲烷/甲醇, V:V=20:1~10:1]进行纯化, 得固体2.
(
S)-11-((苄氧基)甲基)-4-乙基-4-羟基-1,12-二氢- 14
H-吡喃[3',4':6,7]吲哚唑[1,2-
b]喹啉-3,14(4
H)-二酮(
2a): 自由基源为苄氧基乙醛, 经Flash柱纯化[二氯甲烷/甲醇,
V:
V=60:1~40:1], 产物为淡黄色固体粉末(14 mg, 产率34.70%). m.p.<250 ℃ (lit.
[29] 213~215 ℃);
1H NMR (400 MHz, DMSO-
d6)
δ: 8.22 (dd,
J=8.6, 1.4 Hz, 1H), 8.18 (dd,
J=8.6, 1.3 Hz, 1H), 7.86 (ddd,
J=8.3, 6.8, 1.3 Hz, 1H), 7.72 (ddd,
J=8.3, 6.8, 1.3 Hz, 1H), 7.43~7.22 (m, 6H), 6.54 (s, 1H), 5.43 (s, 2H), 5.32 (s, 2H), 5.25 (s, 2H), 4.72 (s, 2H), 1.93~1.83 (m,
J=7.1 Hz, 2H), 0.89 (t,
J=7.3 Hz, 3H);
13C NMR (101 MHz, DMSO
-d6)
δ: 172.98, 157.21, 151.95, 150.45, 148.62, 145.87, 139.67, 138.36, 130.63, 130.18, 128.86, 128.60, 128.52, 128.21, 128.18, 128.13, 126.34, 125.40, 124.86, 119.58, 97.09, 72.87, 72.80, 67.15, 65.75, 50.91, 30.90, 8.27; HRMS (ESI) calcd for C
28H
25N
2O
5 [M+H]
+ 469.17580, found 469.17520.
(S)-11-(溴甲基)-4-乙基-4-羟基-1,12-二氢-14H-吡喃[3',4':6,7]吲哚唑[1,2-b]喹啉-3,14(4H)-二酮(2b): 自由基源为溴代乙醛缩二甲醇, 经Flash柱纯化[二氯甲烷/甲醇, V:V=60:1~40:1], 产物为黄色固体粉末(5 mg, 产率13.16%). m.p.>250 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.40 (dd, J=8.5, 1.4 Hz, 1H), 8.23 (dd, J=8.5, 1.3 Hz, 1H), 7.91 (ddd, J=8.4, 6.8, 1.4 Hz, 1H), 7.82 (ddd, J=8.3, 6.8, 1.3 Hz, 1H), 7.34 (s, 1H), 6.56 (d, J=79.9 Hz, 1H), 5.45 (s, 2H), 5.36 (s, 2H), 5.33 (s, 2H), 1.93~1.81 (m, 2H), 0.89 (t, J=7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 172.96, 157.25, 153.03, 150.46, 149.16, 145.90, 138.89, 131.01, 130.38, 129.37, 128.53, 125.82, 124.56, 119.85, 97.40, 72.85, 65.75, 49.99, 30.73, 25.82, 8.24; MS (ESI) m/z: 441.0 ([M+H]+); HRMS (ESI) calcd for C21H18BrN2O4 [M+H]+ 441.04445, found 441.04338.
(S)-4-乙基-4-羟基-11-(羟甲基)-1,12-二氢-14H-吡喃[3',4':6,7]吲哚唑[1,2-b]喹啉-3,14(4H)-二酮化合物(2e): 自由基源为甲醇, 产物为白色固体(16 mg, 产率49.10%). m.p.>250 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.18 (dd, J=3.9, 1.3 Hz, 1H), 8.16 (dd, J=3.9, 1.2 Hz, 1H), 7.85 (ddd, J=8.4, 6.8, 1.3 Hz, 1H), 7.70 (ddd, J=8.3, 6.8, 1.3 Hz, 1H), 7.34 (s, 1H), 6.54 (s, 1H), 5.84 (t, J=5.4 Hz, 1H), 5.44 (s, 2H), 5.40 (s, 2H), 5.28 (d, J=5.3 Hz, 2H), 1.95~1.81 (m, J=7.1 Hz, 2H), 0.87 (t, J=7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 173.00, 157.30, 153.15, 150.47, 148.45, 145.79, 143.68, 130.51, 130.13, 127.99, 127.34, 125.87, 124.64, 119.46, 96.98, 72.89, 65.75, 59.78, 51.20, 30.71, 8.29; HRMS (ESI) calcd for C21H19N2O5 [M+H]+ 379.12855, found 379.12788.
(S)-4-乙基-4-羟基-11-(2-羟乙基)-1,12-二氢-14H-吡喃[3',4':6,7]吲哚唑[1,2-b]喹啉-3,14(4H)-二酮(2f): 按喜树碱100 mg投料, 自由基源为乙醇, 产物为黄色固体(16 mg, 产率14.20%). m.p.>250 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.24 (dd, J=8.6, 1.3 Hz, 1H), 8.13 (dd, J=8.5, 1.2 Hz, 1H), 7.82 (ddd, J=8.3, 6.7, 1.3 Hz, 1H), 7.68 (ddd, J=8.4, 6.8, 1.4 Hz, 1H), 7.32 (s, 1H), 6.57 (s, 1H), 5.44 (s, 2H), 5.27 (s, 2H), 4.94 (s, 1H), 3.87~3.80 (m, 2H), 3.36 (t, J=6.4 Hz, 2H), 1.88 (q, J=7.0 Hz, 2H), 0.90 (t, J=7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 172.99, 157.27, 151.97, 150.51, 148.89, 146.41, 142.39, 130.34, 130.25, 130.13, 127.83, 127.68, 124.77, 119.39, 97.15, 72.87, 65.74, 61.16, 50.59, 33.34, 30.76, 8.26; MS (ESI) m/z: 393.1 ([M+H]+).
(S)-4,11-二乙基-4-羟基-1,12-二氢-14H-吡喃[3',4':6,7]吲哚唑[2-b]喹啉-3,14(4H)-二酮(2g): 自由基源为乙基硼酸, 经Flash柱纯化[二氯甲烷/甲醇, V:V=60:1~40:1], 产物为黄色固体(8 mg, 产率24.68%). m.p. >250 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.29 (dd, J=8.6, 1.4 Hz, 1H), 8.17 (dd, J=8.4, 1.3 Hz, 1H), 7.86 (ddd, J=8.4, 6.8, 1.4 Hz, 1H), 7.73 (ddd, J=8.3, 6.8, 1.3 Hz, 1H), 7.33 (s, 1H), 6.55 (s, 1H), 5.44 (s, 2H), 5.33 (s, 2H), 3.23 (q, J=7.6 Hz, 2H), 1.90 (qt, J=14.1, 7.2 Hz, 2H), 1.39~1.28 (m, 3H), 0.88 (q, J=7.0 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 173.00, 157.32, 152.45, 150.50, 148.99, 146.51, 146.05, 130.44, 130.41, 128.54, 128.02, 127.06, 124.53, 119.42, 97.10, 72.87, 65.75, 50.02, 30.72, 22.69, 14.45, 8.25; HRMS (ESI) calcd for C22H21- N2O5 [M+H]+ 393.14450, found 393.14360.
将喜树碱(6 g, 17.2 mmol)分散于600 mL水中, 在冰浴下滴加323.9 g浓硫酸, 使喜树碱完全溶解. 在室温下, 向体系中加入FeSO4•7H2O (8.41 g, 30.23 mmol), 搅拌5 min后, 加入丙烯醛(14.48 g, 258.36 mmol). 将体系移入冰浴, 滴加质量分数为30%的H2O2 (7 mL, 68.6 mmol), 过程中控制内温<20 ℃. 反应30 min后补加FeSO4•7H2O (8.41 g, 30.23 mmol), 搅拌5 min后将体系移入冰浴, 滴加质量分数为30%的H2O2(7 mL, 68.6 mmol), 过程中控制内温<20 ℃, 继续反应30 min. 反应结束后加入质量分数为30%的NaOH水溶液淬灭反应, 过滤析出的固体, 水相以EA萃取(200 mL×3), 浓缩后合并所得固体, 经Flash柱纯化[二氯甲烷/甲醇, V:V=20:1~10:1]得黄色固体(3.374 g, 产率49.92%). m.p.>250 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.24 (dd, J=8.6, 1.4 Hz, 1H), 8.13 (dd, J=8.6, 1.3 Hz, 1H), 7.82 (ddd, J=8.3, 6.7, 1.3 Hz, 1H), 7.67 (ddd, J=8.3, 6.8, 1.3 Hz, 1H), 7.32 (s, 1H), 6.56 (s, 1H), 5.44 (s, 2H), 5.25 (s, 2H), 4.93 (t, J=5.6 Hz, 1H), 3.81 (q, J=6.0 Hz, 2H), 3.35 (t, J=6.4 Hz, 2H), 1.94~1.82 (m, J=7.2 Hz, 2H), 0.90 (t, J=7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 172.98, 157.26, 151.96, 150.50, 148.89, 146.40, 142.38, 130.33, 130.25, 130.11, 127.82, 127.67, 124.76, 119.38, 97.14, 72.87, 65.74, 61.16, 50.57, 33.33, 30.78, 8.26; HRMS (ESI) calcd for C22H21N2O5 [M+H]+ 393.14450, found 393.14283.
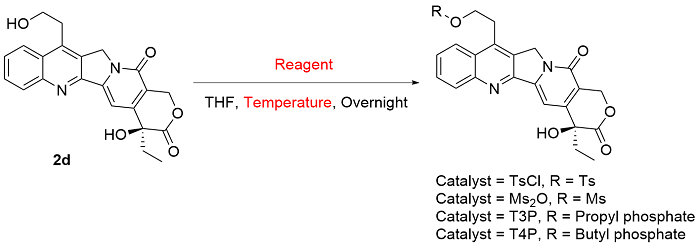
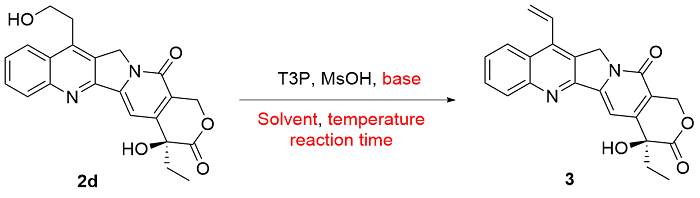
将化合物2d (133 mg, 0.3 mmol)分散于4.39 mL THF中, 向混悬液中加入T3P (667 mg, 1.05 mmol, 50% EA溶液), 室温搅拌5 min后, 加入MsOH (136 μL, 2.10 mmol)和二乙胺(194 μL, 2.39 mmol), 70 ℃反应5 h. 反应结束后加入水淬灭, 过滤析出的黄色固体, 以水, MTBE打浆处理, 过滤得橙色固体(75 mg, 收率68.77%). m.p.>250 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.32 (dd, J=8.6, 1.3 Hz, 1H), 8.20 (dd, J=8.4, 1.2 Hz, 1H), 7.89 (ddd, J=8.4, 6.7, 1.3 Hz, 1H), 7.75 (ddd, J=8.3, 6.8, 1.3 Hz, 1H), 7.58 (dd, J=17.7, 11.7 Hz, 1H), 7.36 (s, 1H), 6.56 (s, 1H), 6.16~6.05 (m, 2H), 5.45 (s, 2H), 5.40 (s, 2H), 1.87 (dq, J=14.0, 7.0 Hz, 2H), 0.89 (t, J=7.3 Hz, 3H); HRMS (ESI) calcd for C22H19N2O4 [M+H]+ 375.13393, found 375.13242.
在3 mL密封反应瓶中加入化合物3 (20 mg, 0.05 mmol)、DMA (0.8 mL)和Tf2NH (6 mg, 0.02 mmol), 搅拌溶解为黄色溶液. 搅拌5 min后加入异丙胺(28 μL, 0.32 mmol), 密封反应瓶, 在60 ℃下反应过夜. 反应结束后反应液经反向纯化[Luna C18, 21.2 mm×250 mm, 10 μm, acetonitrile/trifluoroacetic acid aq. (ϕ=0.05%), V:V=10:90 (5 min)~42:58 (27 min)~90:10 (5 min), 25 ℃, 20 mL/min, 254 nm]得到贝洛替康(11 mg, 纯度99%, 收率47.50%). 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (s, 1H), 8.32 (d, J=8.4 Hz, 1H), 8.21 (d, J=8.4 Hz, 1H), 7.90 (t, J=7.7 Hz, 1H), 7.78 (t, J=7.6 Hz, 1H), 7.36 (s, 1H), 6.58 (s, 1H), 5.46 (s, 2H), 5.40 (s, 2H), 3.56 (s, 2H), 3.39 (dq, J=12.2, 6.0 Hz, 1H), 3.27~3.25 (m, 2H), 1.94~1.83 (m, J=7.2 Hz, 2H), 1.26 (d, J=6.4 Hz, 6H), 0.89 (t, J=7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 172.95, 157.31, 152.53, 150.57, 148.91, 146.21, 139.08, 130.73, 130.50, 130.26, 128.49, 127.20, 124.26, 119.65, 97.31, 72.86, 65.73, 50.39, 50.31, 43.47, 30.77, 26.64, 19.27, 19.23, 8.24; HRMS (ESI) calcd for C25H28N3O4 [M+H]+ 434.20743, found 434.20637.
辅助材料(Supporting Information) 化合物
2a,
2b,
2d~
2g,
3和贝洛替康的
1H NMR,
13C NMR和MS谱图. 这些材料可以免费从本刊网站(
http://sioc-journal.cn/)上下载.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}